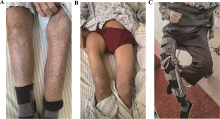
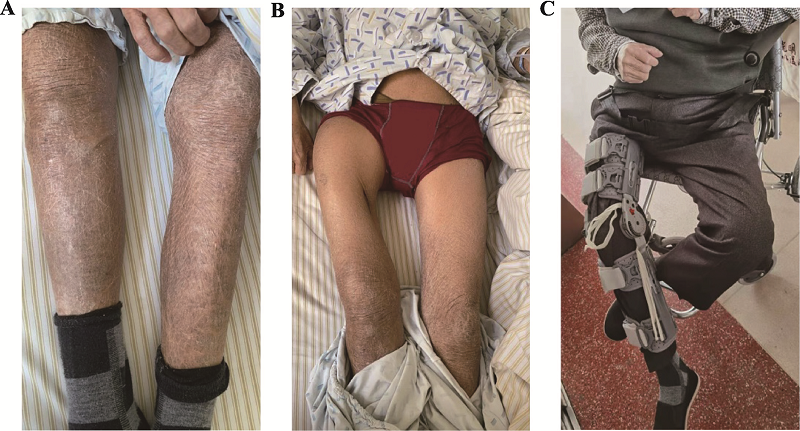
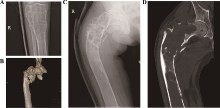
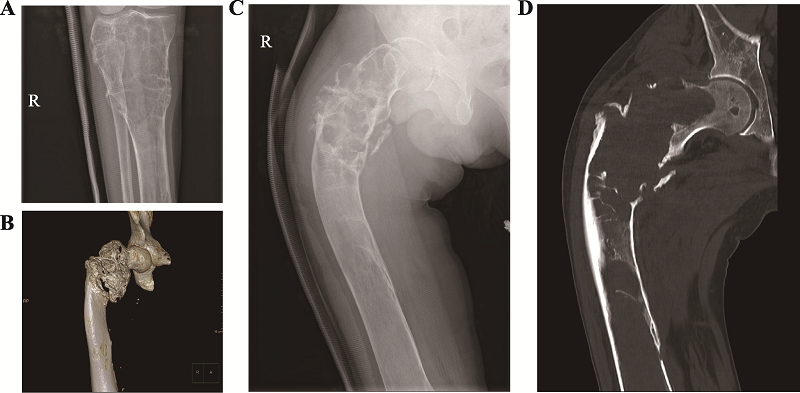
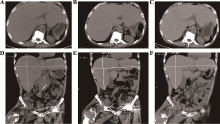
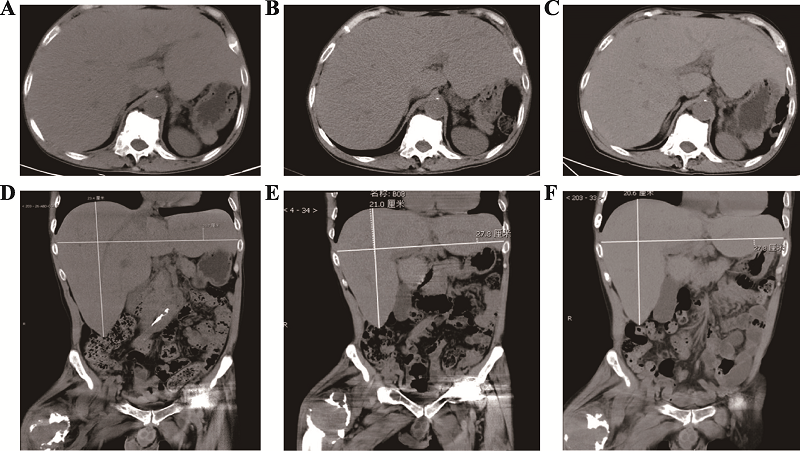
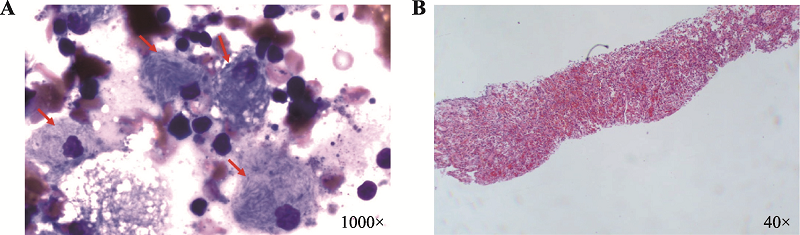
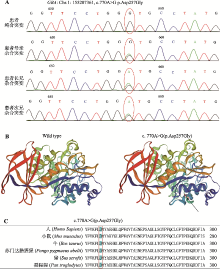
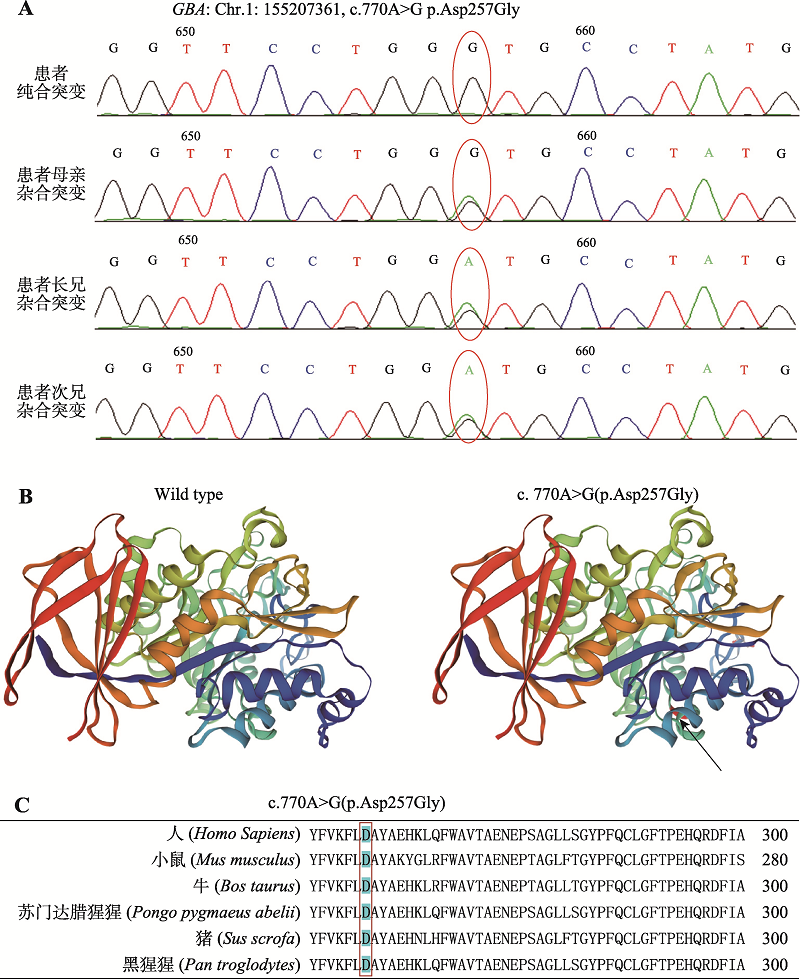
| [1] |
Brady RO, Kanfer JN, Shapiro D. Metabolism of glucocerebrosides. II. Evidence of an enzymatic deficiency in Gaucher's disease. Biochem Biophys Res Commun, 1965, 18: 221-225.
doi: 10.1016/0006-291X(65)90743-6
|
| [2] |
Erythrocyte Diseases Anemia Group, Hematology Branch, Chinese Medical Association. Expert consensus on the diagnosis and treatment of adult Gaucher disease in China (2020). Natl Med J China, 2020, 100(24): 1841-1849.
|
|
中华医学会血液学分会红细胞疾病贫血学组.中国成人戈谢病诊治专家共识(2020). 中华医学杂志, 2020, 100(24): 1841-1849.
|
| [3] |
Riboldi GM, Di Fonzo AB. GBA, Gaucher disease, and Parkinson's disease: from genetic to clinic to new therapeutic approaches. Cells, 2019, 8(4): 364.
doi: 10.3390/cells8040364
|
| [4] |
Zhang WM, Deng LS, Meng Y, Yao FX, Qiu ZQ, Duan YL, Huang SZ, Shi HP. Analysis of gene mutations in Chinese people with Gaucher disease. Natl Med J China, 2009, 89(48): 3397-3400.
|
|
张为民, 邓亮生, 孟岩, 姚凤霞, 邱正庆, 段彦龙, 黄尚志, 施惠平. 中国人戈谢病基因突变的分析. 中华医学杂志, 2009, 89(48): 3397-3400.
|
| [5] |
Park JK, Orvisky E, Tayebi N, Kaneski C, Lamarca ME, Stubblefield BK, Martin BM, Schiffmann R, Sidransky E. Myoclonic epilepsy in Gaucher disease: genotype- phenotype insights from a rare patient subgroup. Pediatr Res, 2003, 53(3): 387-395.
doi: 10.1203/01.PDR.0000049515.79882.94
|
| [6] |
Grabowski GA, Zimran A, Ida H. Gaucher disease types 1 and 3: phenotypic characterization of large populations from the ICGG Gaucher Registry. Am J Hematol, 2015, 90 Suppl 1: S12-S18.
|
| [7] |
Hughes D, Mikosch P, Belmatoug N, Carubbi F, Cox T, Goker-Alpan O, Kindmark A, Mistry P, Poll L, Weinreb N, Deegan P. Gaucher Disease in bone: from pathophysiology to practice. J Bone Miner Res, 2019, 34(6): 996-1013.
doi: 10.1002/jbmr.3734
pmid: 31233632
|
| [8] |
Giraldo P, Pérez-López J, Núñez R, de la Puebla RF, Luño E, Saura-Grau S, Bureo JC, Plaza S, de la Serna J. Patients with type 1 Gaucher disease in Spain: a cross-sectional evaluation of health status. Blood Cells Mol Dis, 2016, 56(1): 23-30.
doi: 10.1016/j.bcmd.2015.10.001
|
| [9] |
Mistry PK, Liu J, Yang M, Nottoli T, McGrath J, Jain D, Zhang K, Keutzer J, Chuang WL, Mehal WZ, Zhao HY, Lin AP, Mane S, Liu X, Peng YZ, Li JH, Agrawal M, Zhu LL, Blair HC, Robinson LJ, Iqbal J, Sun L, Zaidi M. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc Natl Acad Sci USA, 2010, 107(45): 19473-19478.
doi: 10.1073/pnas.1003308107
pmid: 20962279
|
| [10] |
Poll LW, Cox ML, Godehardt E, Steinhof V, vom Dahl S, Whole body MRI in type I Gaucher patients: evaluation of skeletal involvement. Blood Cells Mol Dis, 2011, 46(1): 53-59.
doi: 10.1016/j.bcmd.2010.10.005
|
| [11] |
Kamath RS, Lukina E, Watman N, Dragosky M, Pastores GM, Arreguin EA, Rosenbaum H, Zimran A, Aguzzi R, Puga AC, Norfleet AM, Peterschmitt MJ, Rosenthal DI. Skeletal improvement in patients with Gaucher disease type 1: a phase 2 trial of oral eliglustat. Skeletal Radiol, 2014, 43(10): 1353-1360.
doi: 10.1007/s00256-014-1891-9
pmid: 24816856
|
 ), Hua Yin1, Rong Wang1, Feipeng Zhu2, Jianyong Li1, Ruinan Lu1(
), Hua Yin1, Rong Wang1, Feipeng Zhu2, Jianyong Li1, Ruinan Lu1(