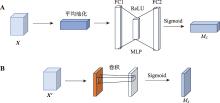
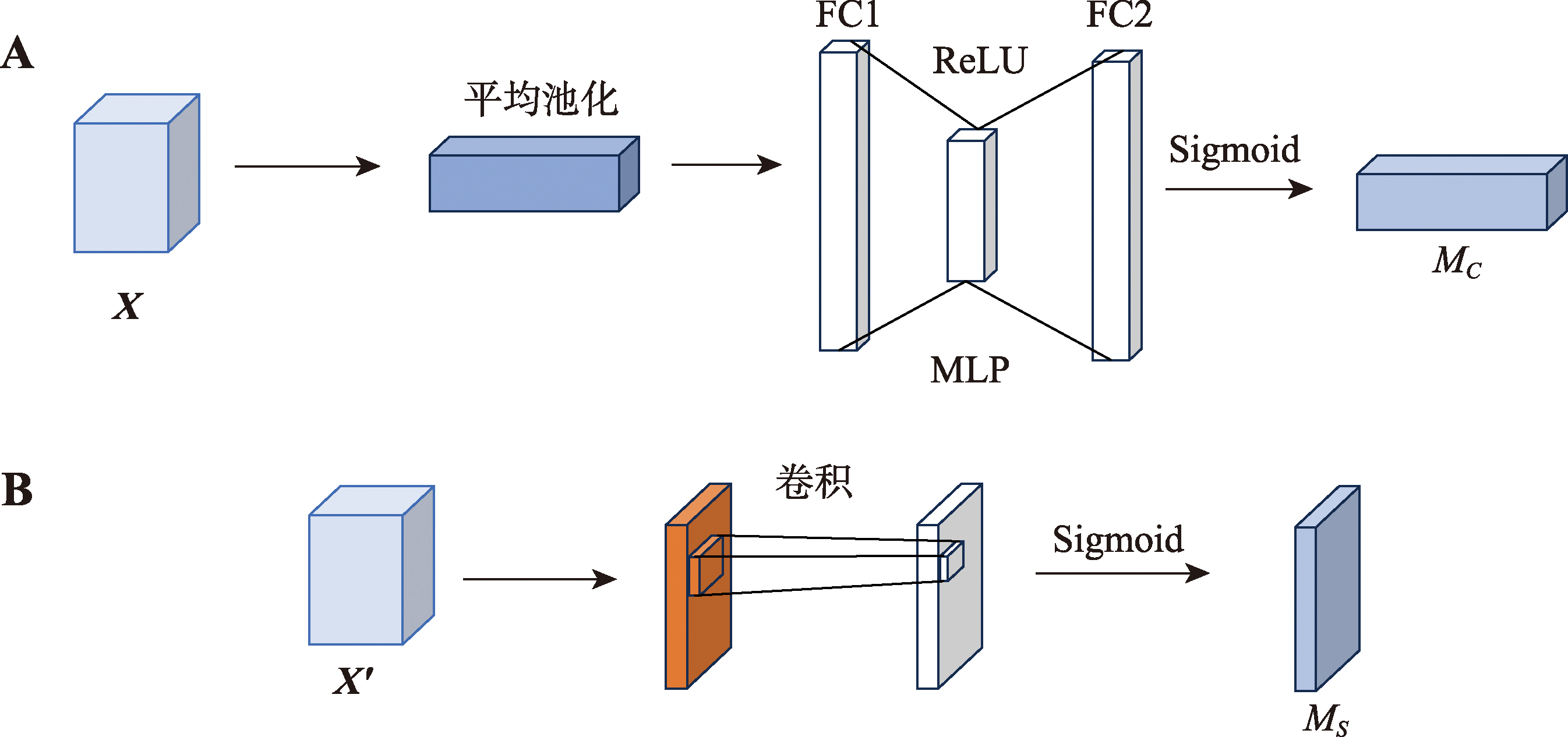
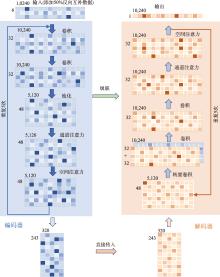
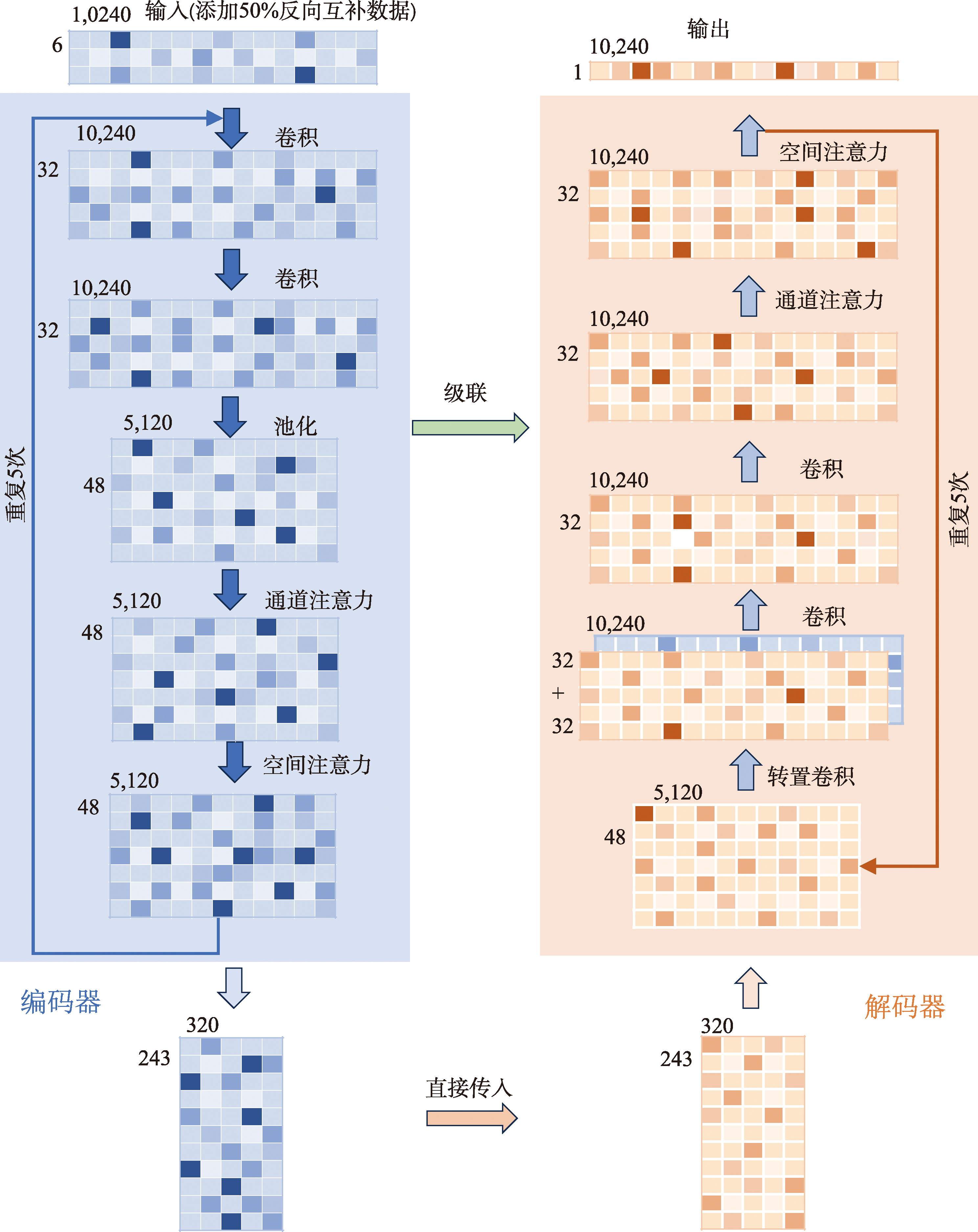
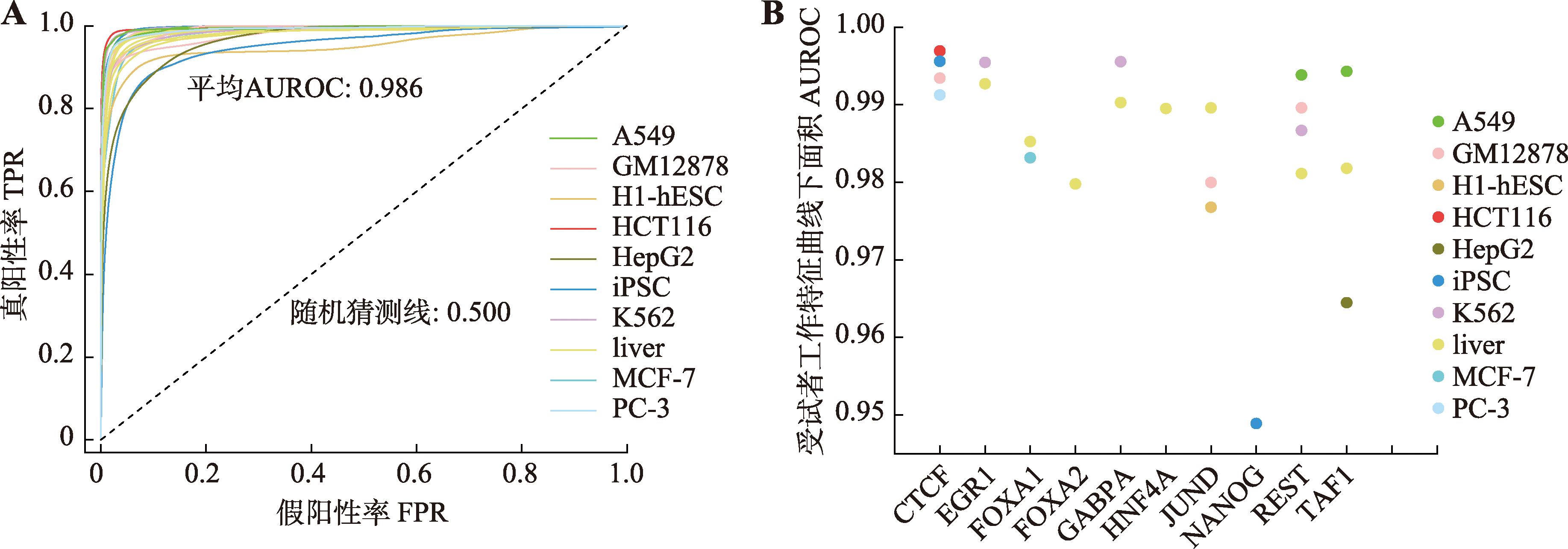
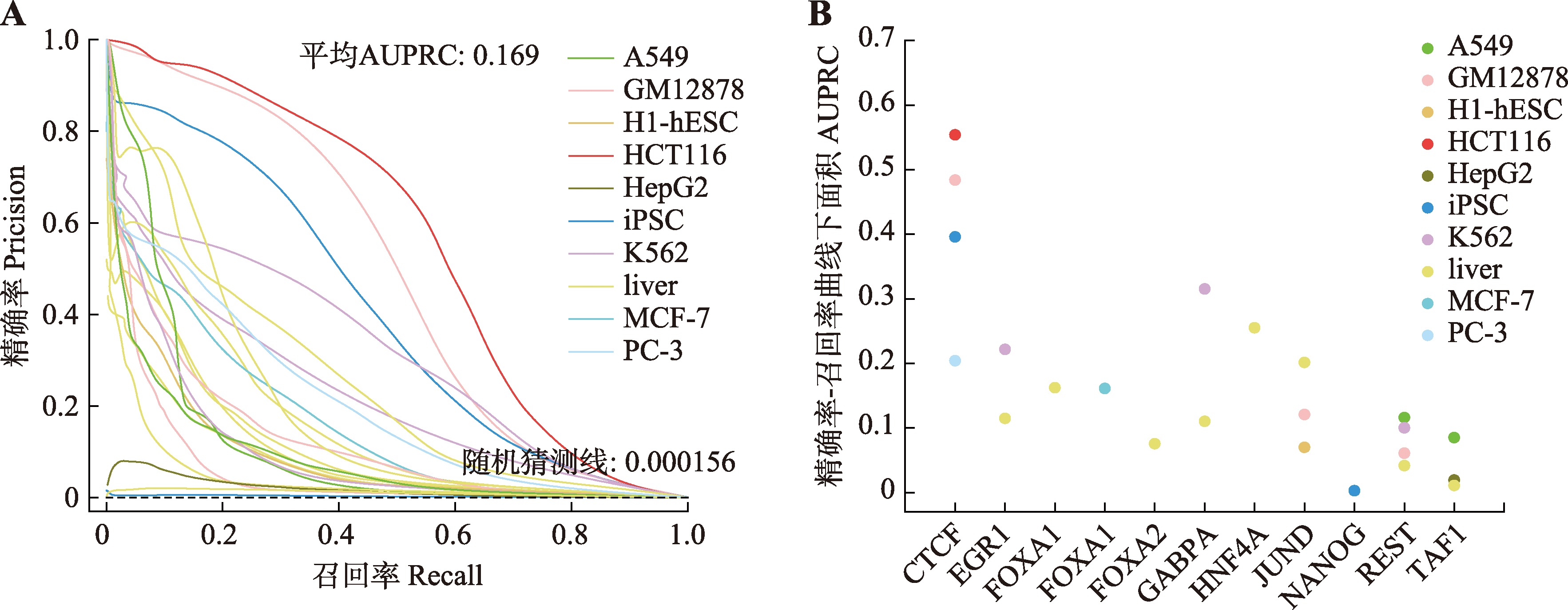
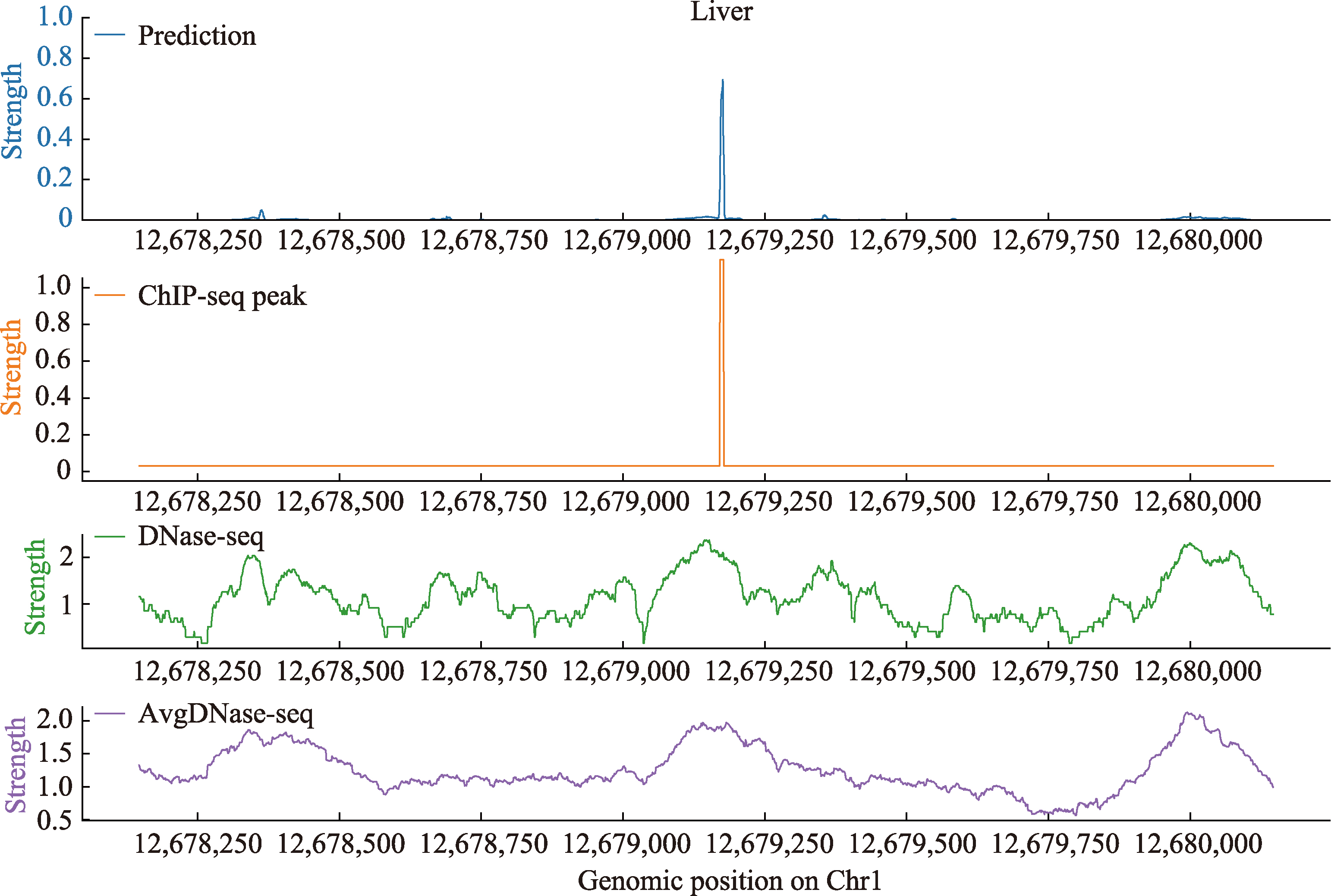
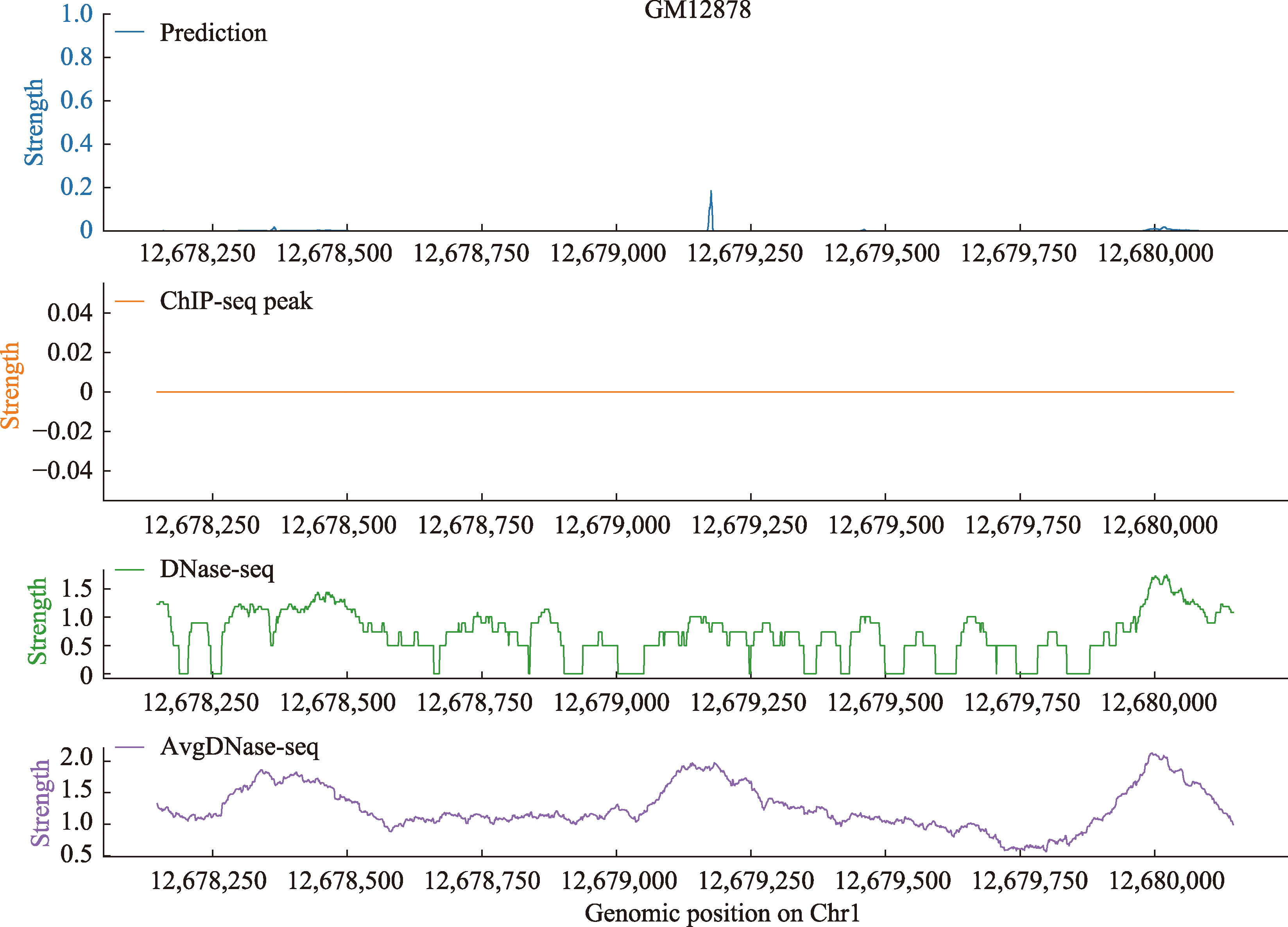
| [1] |
Zhao ZH. Research on prediction of transcription factor binding sites based on deep kernel network[Dissertation]. Beijing Jiaotong University, 2023.
|
|
赵子涵. 基于深度核网络的转录因子结合位点预测的研究[学位论文]. 北京交通大学, 2023.
|
| [2] |
Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei GH, Palin K, Vaquerizas JM, Vincentelli R, Luscombe NM, Hughes TR, Lemaire P, Ukkonen E, Kivioja T, Taipale J. DNA-binding specificities of human transcription factors. Cell, 2013, 152(1): 327-339.
|
| [3] |
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin YM, Albu M, Chen XT, Taipale J, Hughes TR, Weirauch MT. The human transcription factors. Cell, 2018, 172(4): 650-665.
pmid: 29425488
|
| [4] |
Johnson DS, Mortazavi A, Myers RM, Wold B. Genome- wide mapping of in vivo protein-DNA interactions. Science, 2007, 316(5830): 1497-1502.
pmid: 17540862
|
| [5] |
He QY, Johnston J, Zeitlinger J. ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat Biotechnol, 2015, 33(4): 395-401.
pmid: 25751057
|
| [6] |
Zhang ZY, Zhou YP, Meng ZX. The protocol of CUT&Tag for metabolic tissue cells. Hereditas(Beijing), 2022, 44(10): 958-966.
|
|
张子寅, 周燕萍, 孟卓贤. CUT&Tag技术在代谢组织细胞的实验操作. 遗传, 2022, 44(10): 958-966.
|
| [7] |
Hesselberth JR, Chen XY, Zhang ZH, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, Fields S, Stamatoyannopoulos JA. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods, 2009, 6(4): 283-289.
pmid: 19305407
|
| [8] |
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA- binding proteins and nucleosome position. Nat Methods, 2013, 10(12): 1213-1218.
pmid: 24097267
|
| [9] |
Suresh SK. Beginner’s guide to investigating protein: DNA interactions using electrophoretic mobility shift assays (EMSAs). Biochem (Lond), 2024, 46(5): 8-11.
|
| [10] |
Hook H, Zhao RW, Bray D, Keenan JL, Siggers T. High- throughput analysis of the cell and DNA site-specific binding of native NF-κB dimers using nuclear extract protein- binding microarrays (nextPBMs). Methods Mol Biol, 2021, 2366: 43-66.
pmid: 34236632
|
| [11] |
Zou J, Huss M, Abid A, Mohammadi P, Torkamani A, Telenti A. A primer on deep learning in genomics. Nat Genet, 2019, 51(1): 12-18.
pmid: 30478442
|
| [12] |
Ghandi M, Lee D, Mohammad-Noori M, Beer MA. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol, 2014, 10(7): e1003711.
pmid: 25033408
|
| [13] |
Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, Pritchard JK. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res, 2011, 21(3): 447-455.
pmid: 21106904
|
| [14] |
Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods, 2015, 12(10): 931-934.
pmid: 26301843
|
| [15] |
Alipanahi B, Delong A, Weirauch MT, Frey BJ. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol, 2015, 33(8): 831-838.
pmid: 26213851
|
| [16] |
Quang D, Xie XH. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res, 2016, 44(11): e107.
pmid: 27084946
|
| [17] |
Wang M, Tai C, E W, Wei LP. DeFine: deep convolutional neural networks accurately quantify intensities of transcription factor-DNA binding and facilitate evaluation of functional non-coding variants. Nucleic Acids Res, 2018, 46(11): e69.
pmid: 29617928
|
| [18] |
Avsec Ž, Weilert M, Shrikumar A, Krueger S, Alexandari A, Dalal K, Fropf R, McAnany C, Gagneur J, Kundaje A, Zeitlinger J. Base-resolution models of transcription- factor binding reveal soft motif syntax. Nat Genet, 2021, 53(3): 354-366.
pmid: 33603233
|
| [19] |
Li HY, Quang D, Guan YF. Anchor: trans-cell type prediction of transcription factor binding sites. Genome Res, 2019, 29(2): 281-292.
pmid: 30567711
|
| [20] |
Quang D, Xie XH. FactorNet: a deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods, 2019, 166: 40-47.
pmid: 30922998
|
| [21] |
Li HY, Guan YF. Fast decoding cell type-specific transcription factor binding landscape at single-nucleotide resolution. Genome Res, 2021, 31(4): 721-731.
pmid: 33741685
|
| [22] |
Fu LY, Zhang LH, Dollinger E, Peng QK, Nie Q, Xie XH. Predicting transcription factor binding in single cells through deep learning. Sci Adv, 2020, 6(51): eaba9031.
pmid: 33355120
|
| [23] |
Yang TQ, Henao R. TAMC: a deep-learning approach to predict motif-centric transcriptional factor binding activity based on ATAC-seq profile. PLoS Comput Biol, 2022, 18(9): e1009921.
pmid: 36094959
|
| [24] |
Luong MT, Pham H, Manning CD. Effective approaches to attention-based neural machine translation. arXiv preprint arXiv, 2015, 1508.04025.
|
| [25] |
Wang YQ, Huang ML, Zhao L, Zhu XY. Attention-based LSTM for aspect-level sentiment classification. Proceedings of the 2016 conference on empirical methods in natural language processing, 2016: 606-615.
|
| [26] |
Chen C, Hou J, Shi XW, Yang H, Birchler JA, Cheng JL. DeepGRN: prediction of transcription factor binding site across cell-types using attention-based deep neural networks. BMC Bioinformatics, 2021, 22(1): 38.
pmid: 33522898
|
| [27] |
Yao Z, Zhang WJ, Song P, Hu YX, Liu JX. DeepFormer: a hybrid network based on convolutional neural network and flow-attention mechanism for identifying the function of DNA sequences. Brief Bioinform, 2023, 24(2): bbad095.
pmid: 36917472
|
| [28] |
Ronneberger O, Fischer P, Brox T. U-net:Convolutional networks for biomedical image segmentation. In: International Conference on Medical Image Computing and Computer-Assisted Intervention. Cham: Springer International Publishing, 2015: 234-241.
|
| [29] |
Keilwagen J, Posch S, Grau J. Accurate prediction of cell type-specific transcription factor binding. Genome Biol, 2019, 20(1): 9.
pmid: 30630522
|
| [30] |
Hu DD, Zhang ZT, Niu GC. Lane line detection incurporating CBAM mechanism and deformable convolutional network. Journal of Beijing University of Aeronautics and Astronautics, 2024, 50(7): 2150-2160.
|
|
胡丹丹, 张忠婷, 牛国臣. 融合CBAM注意力机制与可变形卷积的车道线检测. 北京航空航天大学学报, 2024, 50(7): 2150-2160.
|
| [31] |
Johnson OV, Chew XY, Khaw KW, Lee MH. ps-CALR: periodic-shift cosine annealing learning rate for deep neural networks. IEEE Access, 2023, 11: 139171-139186.
|
| [32] |
Li FF, Wang Y, Gu JH, Zhang YM, Liu FS, Ni ZH. E2F family play important roles in tumorigenesis. Hereditas(Beijing), 2023, 45(7): 580-592.
|
|
李飞飞, 王韵, 顾冀海, 张玉明, 柳峰松, 倪志华. E2F家族转录因子在肿瘤发生中的作用. 遗传, 2023, 45(7): 580-592.
|
| [33] |
Fan XD, Zhou J, Jiang XL, Xin MZ, Hou LM. CSAP- UNet: convolution and self-attention paralleling network for medical image segmentation with edge enhancement. Comput Biol Med, 2024, 172: 108265.
pmid: 38461698
|

 ), 陈忠兴1,2, 康琦林1,2, 李龙飞1,2, 杨佳慧1,2, 张雨亭1,2
), 陈忠兴1,2, 康琦林1,2, 李龙飞1,2, 杨佳慧1,2, 张雨亭1,2