


植物古基因组学是一门从现存物种中重建和分析其祖先基因组的学科,旨在研究植物祖先基因组的进化历史[1,2]。随着测序技术不断革新,测序成本不断降低,目前已公布了180多种植物的参考基因组数据[3],其中甜橙(Citrus sinensis)[4]、黄瓜(Cucumis sativus)[5]和巨桉(Eucalyptus grandis)[6]等植物基因组组装到染色体水平;海草(Zostera marina)[7]、挪威云杉(Picea abies)[8]和毛竹(Phyllostachys heterocycla)[9]等植物基因组组装到Scaffold水平,这为古基因组学研究提供了大量有价值的序列数据资源。古基因组学计算依赖于序列测序准确度和组装水平,低质量的序列会影响古基因组学计算的准确度。如何提高基因组测序和组装的准确度和长度一直是本研究领域所关注的热点。
近年来,大批量植物基因组测序和重测序项目推动了古基因组学的发展,提出的全基因组复制(whole-genome duplication,WGD)(或古多倍化)事件是植物基因产生新功能化的主要进化推动力[10,11],为植物适应基因重复提供原始材料,有益于植物适应生理和遗传改变[12]。有研究报道,通过古基因组学研究可溯源到由7931条原基因模型和7条染色体构成的双子叶植物祖先基因组,以及由9138条原基因模型和5条染色体构成的单子叶植物祖先基因组[1]。而在裸子植物进化史中,WGD事件使松柏类植物与其他裸子植物分化开来[13],银杏除与被子植物共享一次WGD事件外,近期发生过一次银杏特异性WGD事件[14]。由于裸子植物基因组数据有限,裸子植物祖先基因组基因模型尚未确定。
参考基因组序列是古基因组学研究的重要资源,植物全基因组复制事件鉴定是古基因组学研究的重要途径之一。本文主要综述了植物基因组测序与组装研究进展、植物古基因组学的原理、WGD事件研究历程和鉴定方法及其对植物基因组进化的作用、植物祖先基因组进化场景,并对未来古基因组学研究进行了展望。
1 植物基因组测序与组装
基因组(genome)是指一个物种单倍体的染色体数目及其所携带的全部基因,包括每个染色体的序列加上细胞器中的任何DNA,本文主要指植物核基因组。植物基因组通常具有重复序列多、多倍化和杂合度高等特征,使基因组组装复杂化[15,16]。由于Sanger、454、SOLid和Illumina测序技术读长短,不能有效地组装基因组复杂重复区域,特别是非自交或重排杂合基因组[17]。随着高通量测序技术的发展,第三代单分子实时测序(single-molecule real-time sequencing, PacBio)技术使基因组测序读长更长,可以辅助组装出高质量的植物基因组[3]。近年来,已综合运用PacBio和其他测序技术成功组装了凤梨(Ananas comosus)、欧洲白桦(Betula pendula)、芥菜(Brassica juncea)、木豆(Cajanus cajan)、藜麦(Chenopodium quinoa)、一年生辣椒(Cpsicum annuum)、中华辣椒(C. chinense)、复活草(Oropetium thomaeum)、向日葵(Helianthus annuus)、大麦(Hordeum vulgare)、报春花(Primula veris)、海带(Saccharina japonica)、丹参(Salvia miltiorrhiza)和丝叶狸藻(Utricularia gibba)等植物基因组(表1)。
目前,植物基因组测序项目多采用联合使用多种测序技术的策略,相比单一测序途径具有多种优势。主要表现在:(1)克服高杂合度。例如,凤梨属于自交不亲和、杂合度较高的经济作物。凤梨栽培种‘F153’(A. comosus ‘F153’)基因组存在较高杂合度(1%~2%),发现其k-mer深度频率统计分布中存在2个明显的峰:位于约110层深度处的纯合峰和位于约220层深度处的杂合峰[18]。为克服高杂合度,对F153和其近缘种(A. bracteatus)杂交F1代进行测序,并综合运用454、Illumina和PacBio测序技术,组装出高质量(Scaffold N50 高达11.8 Mb)凤梨基因组;(2)克服复杂倍性。例如,芥菜是Brassica属异源多倍体作物,基因组k-mer频率分布图左峰略微突起,表明其杂合度较低,但复杂倍性使其基因组组装更加困难[19]。Yang等[19]综合运用Illumina和PacBio技术对芥菜基因组进行组装,基因组Scaffold N50高达855 kb;(3)克服高比例重复序列。例如,复活草基因组是已知草类最小的基因组,其重复序列比例显著高于其他草类物种[15]。VanBuren等[15]结合Illumina和PacBio测序技术组装得到复活草基因组,Scaffold N50高达7.1 Mb,组装效果好。复活草基因组较低的杂合度(0.087%)可能与基因组的高连续性相关。向日葵基因组3/4以上的序列是重复序列,组装难度大[20]。Badouin等[20]使用PacBio技术成功组装出3.6 Gb向日葵参考基因组序列。大麦基因组较大(4.79 Gb),而且80.8%的序列属于重复序列,组装难度大,联合使用454、Illumina Hiseq和PacBio测序技术提高了Scaffold长度和精准度,Scaffold N50长达1.9 Mb[21];(4)克服祖先基因组倍性复杂且基因组较大的问题。例如,栽培花生(Arachis hypogaea)是一个异源四倍体(AABB),由二倍体花生A. duranensis (AA)和A. ipaensis (BB)杂交及多倍化而来。栽培花生基因组较大(约2.7 Gb),重复序列比例高达64%,基因组组装难度大。经多倍化后,栽培花生亚基因组A(1.25 Gb)和亚基因组B(1.56 Gb)改变较小,可以通过两个亚基因组组装成完整基因组,运用多种测序方法对A. duranensis和A. ipaensis基因组进行测序,构建了高质量的栽培花生祖先基因组草图[22]。对凤梨、芥菜、大麦、向日葵、复活草和花生等基因组的组装表明,联合应用多种测序技术有助于提高基因组的组装效果,提供更可靠的参考基因组序列。
表1 部分植物基因组组装情况
Table 1
| 物种 | 测序技术 | 基因组大小 | 重复序列比例(%) | 倍性 | 杂合度(%) | N50 (kb) Scaffold/Contig | 文献 |
|---|---|---|---|---|---|---|---|
| 凤梨 | 454+Illumina Hiseq+PacBio | 382 Mb | 44 | 二倍体 | 1~2 | 11800/126.5 | [18] |
| 芥菜 | Illumina Hiseq+PacBio | 955 Mb | 33 | 异源四倍体 | ― | 855/61.3 | [19] |
| 花生基因组A亚组 | Sanger+454+Illumina Hiseq+ PacBio | 1.25 Gb | 61 | 二倍体 | ― | 948/222.93 | [22] |
| 花生基因组B亚组 | 1.56 Gb | 68 | 5343.3/234.92 | ||||
| 巨桉 | Sanger+Illumina | 640 Mb | 50 | 二倍体 | ― | 5000/2261 | [6] |
| 一年生辣椒 | Sanger+454+Illumina Hiseq+ Illumina GA | 3.06 Gb | 76 | 二倍体 | 0.005 | 2470/30.0 | [23] |
| 中华辣椒 | 79 | ||||||
| 木豆 | Sanger+Illumina Hiseq+Illumina GA | 605.78 Mb | 51 | 二倍体 | ― | 516/21.9 | [24] |
| 丝叶狸藻 | PacBio | 101.95 Mb | 58 | 二倍体 | ― | 3424.836 | [25] |
| 欧洲白桦 | 454+SOLid+Illumina MiSeq+PacBio | 440 Mb | 49.23 | 二倍体 | ― | 527.7 | [26] |
| 藜麦 | Illumina Hiseq+PacBio | 1.39 Gb | 64 | 二倍体 | ― | 3846.917 | [27] |
| 大麦 | 454+Illumina Hiseq+PacBio | 4.79 Gb | 80.8 | 二倍体 | ― | 1900/79 | [21] |
| 向日葵 | Sanger+PacBio | 3.6 Gb | 41.2 | 二倍体 | ― | 13.7 | [20] |
| 复活草 | Illumina Hiseq+PacBio | 245 Mb | 43 | 二倍体 | 0.087 | 7100/2400 | [15] |
| 报春花 | Illumina Hiseq+Illumina Miseq+ PacBio | 301.8 Mb | 7 | 二倍体 | ― | 164/9.5 | [28] |
| 海带 | 454+Illumina Hiseq+PacBio | 537 Mb | 40 | 二倍体 | ― | 252/58.9 | [29] |
| 丹参 | 454+Illumina Hiseq+PacBio | 558 Mb | 54 | 二倍体 | 0.003 | 51/12.4 | [30] |
2 植物古基因组学
古基因组学(paleogenomics)最有可能的进化场景主要基于以下假想推断[31]:(1)基于现存物种间直系同源基因重复等事件追溯祖先基因组;(2)基于插入、删除、融合、分裂、易位等事件的最小数量推断从祖先基因组到现存核型的进化历史。其中AL(比对长度AL = ∑高分基因对长度)、CIP(CIP= ∑(高分基因对同源率/AL)×100)和CALP(CALP=AL/长度CDS)是古基因组学计算所需的关键参数[31]。目前,在基于进化关系的比较基因组学中广泛使用经典的CIP/CALP阈值,该阈值等于70%则代表亲缘关系较近的物种基因组间共同祖先可以追溯到50百万年前(million year ago, MYA)以内,该阈值等于50%则代表分化较远的物种基因组间共同祖先可以追溯到50 MYA以外。Salse等[2]提出“三步法”构建祖先基因组,即分析基因组内共线性,鉴定系列特异性基因组内改变事件,推断共有基因组改变事件。不断更新的植物参考基因组序列数据为古基因组学研究提供了更多资源,Murat等[32]对显花植物祖先基因组进化模式进行分析,构建出由7010条原基因模型和7条染色体构成的草类祖先核型、由6284条原基因模型和7条染色体构成的双子叶植物祖先核型、由6707条原基因模型和5条染色体构成的单子叶植物祖先核型以及由1175条原基因模型和15条染色体构成的显花植物最近共同祖先核型,首次系统推断了被子植物祖先基因组的进化史。
3 植物全基因组复制事件
3.1 WGD研究历程
研究植物古基因组学的关键途径是确定WGD事件。1970年,Ohno[33]首次提出多倍化即全基因组复制的假想,阐释二倍体基因组通过WGD进化为四倍体的途径。1997年,Wolfe等[34]在酿酒酵母(Saccharomyces cerevisiae)基因组中证实了WGD假想。2000年,首例植物全基因组草图测序完成,解析了拟南芥(Arabidopsis thaliana)基因组进化历史,提出拟南芥可能有一个四倍化祖先[35]。2007年,葡萄(Vitis vinifera)基因组测序完成,古基因组学研究表明葡萄的祖先基因组可能是古六倍体[36]。对葡萄基因组重复事件的研究奠定了双子叶植物祖先基因组共享六倍化的理论基础,并开启了被子植物祖先基因组进化历史研究的大门。2008年,小立碗藓(Physcomitrella patens)基因组测序完成。通过古基因组学研究发现,陆地植物最近的共同祖先基因组在多倍化后发生重要基因功能分化,如(1)丢失适应水生环境、动力蛋白运输相关基因;(2)获得转运能力、信号转导、抗非生物胁迫相关基因;(3)整体提高基因家族复杂度,这为理解陆地植物进化提供了可靠资源[37]。2011年,卷柏(Selaginella moellendorffii)基因组测序完成,虽然通过比较基因组学没有发现古多倍化证据,但为理解陆地植物基因组进化提供了重要资源[38]。2013年,无油樟(Amborella trichopoda)基因组测序草图公布,WGD分析表明其分化发生在古六倍化前,这为理解被子植物基因组和基因进化提供了重要参考[39]。2015年,凤梨基因组测序完成,古基因组学研究表明其基因组发生了2次WGD事件,染色体重排少,并且保留了单子叶植物祖先基因组多倍化后28条染色体中的25条核型,是保守的单子叶植物参考基因组[18]。2016年,银杏基因组草图完成,古基因组学研究表明银杏发生了2次WGD事件,其中较近1次是银杏特异性WGD[14]。2017年,向日葵基因组测序完成,古基因组学研究表明其经历了3次WGD事件:29 MYA发生的太阳花种系特异性WGD,38~50 MYA菊分支(Asterids)Ⅱ物种共有的WGD,以及122~164 MYA双子叶植物共有的古六倍化,为研究菊分支物种古基因组学进化场景提供了依据[20]。越来越多的植物基因组测序完成,这为古基因组学研究提供了大量资源(图1)。显花植物进化研究表明(图1),被子植物基因组经历了二倍化(α)、四倍化(β)和六倍化(γ)3次古WGD事件,禾本目谷类植物基因组经历了ρ、τ和σ 3次WGD事件[40],植物ρ事件发生在禾本目(Poales)内95~ 115 MYA,小麦、玉米和水稻分化之前,草类和凤梨分化之后[18]。
图1
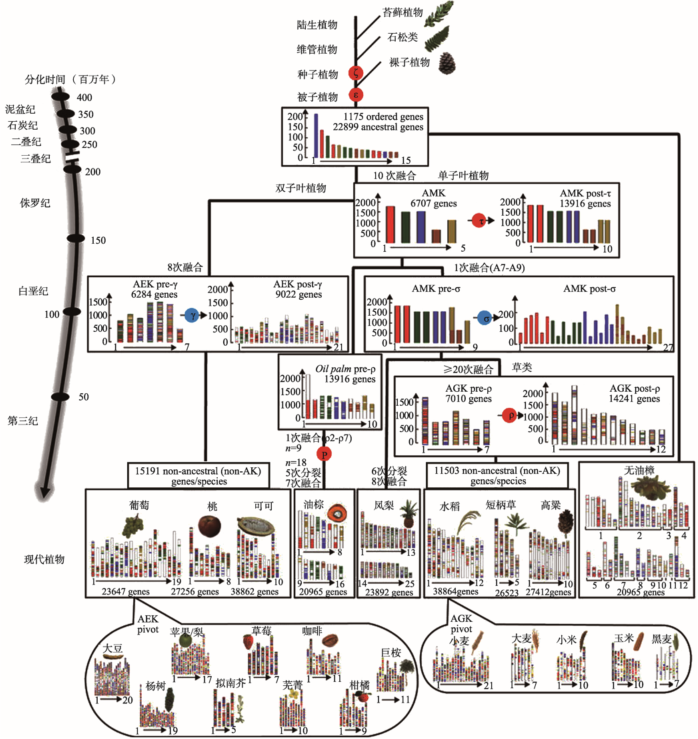
3.2 WGD鉴定方法
目前,主要通过基因组共线性分析、旁系同源基因Ks峰值分布和祖先基因系统发育3种证据链鉴定基因组WGD事件。通过基因组共线性分析发现基因组内存在大量共线区域,是古WGD事件存在的直接证据[18,41],可以通过SynMap(https://genomevolution. org/coge/SynMap.pl)鉴定基因组内部共线性区域及深度和基因组间共线性区域[42]。例如,从凤梨基因组内部鉴定出的388个共线性区域包含64%注释的基因,并分布在25个连锁群,表明凤梨基因组进化历史中存在WGD事件;共线深度分析发现35%基因组存在不止1处重复区域,说明在凤梨系谱中发生了多次WGD事件[18]。在基因组间共线性分析中,无油樟∶葡萄(1∶3)共线性区域定位到古六倍化事件,说明检测到的WGD事件是2种物种共有的。基因组旁系同源基因Ks分布分析是鉴定WGD事件的一种常用途径[14,43],先通过Orthomcl聚类获取旁系同源基因对,然后依据Ks计算值绘制分布图。WGD会导致基因重复峰值的产生,最后通过识别峰值确定WGD事件。但是,串联重复基因积累会影响Ks峰值分布。通过旁系同源基因Ks分布分析已成功在毛竹[9]、鹰嘴豆(Cicer arietinum)[44]、大豆(Glycine max)[45]、小兰屿蝴蝶兰(Phalaenopsis equestris)[46]和银杏[14]等物种基因组中鉴定出WGD事件。祖先基因系统发育分析也是一种鉴定WGD事件的有效途径,即通过系统发育分析识别祖先重复节点基因,进而推断分化时间,再通过祖先基因组重复基因分化时间分布峰值图判断物种间WGD事件[39,46]。通过系统发育研究得到414个无油樟和其他被子植物祖先基因共有节点,其中62%节点构成第一个峰,发生在244 MYA,38%节点构成第二个峰,发生在341 MYA[39]。
3.3 WGD在植物进化中的作用
此外,WGD还为植物适应基因重复的形成提供了原始材料,多倍体比二倍体表现出更强的离子吸收能力和抗逆能力,这种生理关联有益于植物适应新环境或具有挑战性的环境,同等条件下多倍体还会增加种群内适应的速度[12]。同源多倍体来源于种内WGD事件,异源多倍体来源于种间杂交[49]。芸薹属(Brassica)植物经历了3次基因组WGD事件(α、β和γ)[50,51]和种系全基因组三倍化事件[19],是基因组多倍化研究的重要模式物种。海岸红杉(Sequoia sempervirens)是裸子植物中罕见的同源六倍体,是研究裸子植物多倍化的重要材料。海岸红杉基因组WGD事件表明:裸子植物稀有的多倍化可能是由该分支缓慢的二倍化导致的[52]。
4 植物祖先基因组进化
图1描绘了被子植物祖先基因组的进化历程,双子叶植物祖先基因组有7条染色体,单子叶植物祖先基因组有5条染色体,草类祖先基因组有7条染色体,被子植物祖先基因组有15条染色体[32]。和大多数被子植物中检测到的与被子植物家族或更小分支特异性相关的古多倍化事件相比,大多数双子叶植物祖先基因组在进化史中发生了更早的(130~ 150 MYA)古六倍化事件[39]。对西瓜(Citrullus lanatus)古基因组学研究发现,在其基因组进化中发生了7次三倍化,从双子叶植物祖先基因组7条染色体经历了81次分裂和91次融合才形成现存西瓜的11条染色体[53]。凤梨基因组对研究单子叶植物祖先基因组进化具有重要作用,染色体核型进化表明其进化路线开始于单子叶植物祖先基因组的5条染色体,经τ事件后先出现10条染色体后减到9条染色体,经σ事件后出现27条染色体,然后发生8次融合和6次分裂,最终被整合到现存的25条染色体中[32]。复活草基因组和其他草类基因组共同经历了ρ WGD事件[15],二穗短柄草与小麦、水稻、高粱分化时间分别为32~39 MYA、40~53 MYA、45~60 MYA,其基因组内部重复分化时间发生在56~72 MYA,早于草类分化时间[54]。
整合银杏基因组学[14]和古生物学[55]研究表明,银杏可能起源于种子蕨类植物,其基因组经历了2次WGD事件,先后发生在515~735 MYA和74~ 147 MYA。被子植物共有银杏较古老的一次WGD事件,较近的一次可能是银杏特异性WGD事件(图2)。松柏类植物是裸子植物中庞大的一个类群,具有较大的基因组(20~40 Gb,显著大于银杏基因组10.61 Gb),约300 MYA起源于古老的种子植物[56]。Li等[13]对22种裸子植物进行转录组分析,发现裸子植物基因组经历了3次WGD事件,其中2次位于松柏类植物分支(图2)。在裸子植物进化史中,WGD事件使松柏类植物与其他裸子植物分化开,松科和柏科均独自经历1次WGD事件。海岸红杉是柏科中现存的唯一的自然六倍体物种,对其基因组WGD事件进行分析,发现两次WGD事件,先后发生在1.5~10 MYA和0.4~3 MYA[52]。
图2
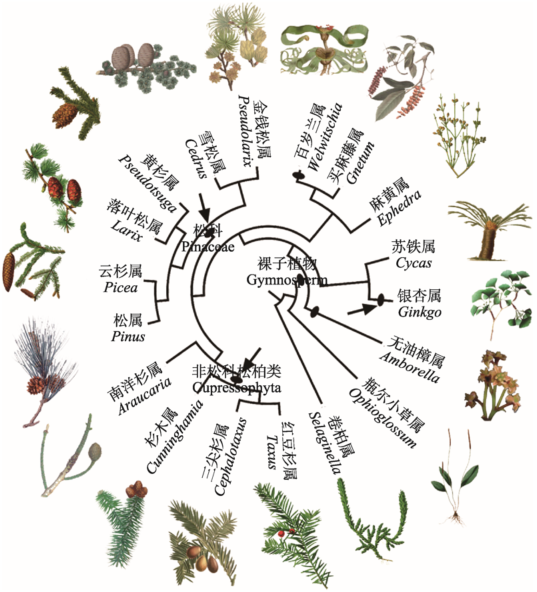
5 展 望
基因组多倍化使植物基因组快速重组,丢失大量基因,增加结构变异[57],是植物基因组进化的重要动力,有利于植物适应新的环境[12,49,58,59]。WGD事件使被子植物基因组大小增加,但平均基因组大小与倍性并不相关[60]。WGD事件发生后,常发生大量同源基因对丢失[61],使鉴定古老的WGD极其困难。通过植物进化重建WGD使推断基因组共线数据更加困难,尤其是使用组装不完整基因组序列检测物种间共线性关系。测序技术在不断革新,测序长度越来越长,准确度和完整度逐步提高,综合运用二代和三代测序平台将为植物古基因组学研究提供更可靠的数据。目前,已有多个组学大数据管理平台储存和提供可靠的植物全基因组序列资源,如JGI(
参考文献
Palaeogenomics of plants: synteny-based modelling of extinct ancestors.

In the past ten years, international initiatives have led to the development of large sets of genomic resources that allow comparative genomic studies between plant genomes at a high level of resolution. Comparison of map-based genomic sequences revealed shared intra-genomic duplications, providing new insights into the evolution of flowering plant genomes from common ancestors. Plant genomes can be presented as concentric circles, providing a new reference for plant chromosome evolutionary relationships and an efficient tool for gene annotation and cross-genome markers development. Recent palaeogenomic data demonstrate that whole-genome duplications have provided a motor for the evolutionary success of flowering plants over the last 50-70 million years.
Improved criteria and comparative genomics tool provide new insights into grass paleogenomics.
In the past decade, a number of bioinformatics tools have been developed to perform comparative genomics studies in plants and animals. However, most of the publicly available and user friendly tools lack common standards for the identification of robust orthologous relationships between genomes leading non-specialists to often over interpret the results of large scale comparative sequence analyses. Recently, we have established a number of improved parameters and tools to define significant relationships between genomes as a basis to develop paleogenomics studies in grasses. Here, we describe our approaches and propose the development of community-based standards that can be used in comparative genomic studies to (i) identify robust sets of orthologous gene pairs, (ii) derive complete sets of chromosome to chromosome relationships within and between genomes and (iii) model common paleo-ancestor genome structures. The rice and sorghum genome sequences are used to exemplify step-by-step a methodology that should allow users to perform accurate comparative genome analyses in their favourite species. Finally, we describe two applications for accurate gene annotation and synteny-based cloning of agronomically important traits.
The impact of third generation genomic technologies on plant genome assembly.
Abstract Since the introduction of next generation sequencing, plant genome assembly projects do not need to rely on dedicated research facilities or community-wide consortia anymore, even individual research groups can sequence and assemble the genomes they are interested in. However, such assemblies are typically not based on the entire breadth of genomic technologies including genetic and physical maps and their contiguities tend to be low compared to the full-length gold standard reference sequences. Recently emerging third generation genomic technologies like long-read sequencing or optical mapping promise to bridge this quality gap and enable simple and cost-effective solutions for chromosomal-level assemblies. Copyright 脗漏 2017 The Authors. Published by Elsevier Ltd.. All rights reserved.
The draft genome of sweet orange (Citrus sinensis).
The genome of the cucumber,Cucumis sativus L.
The genome ofEucalyptus grandis.
The genome of the seagrassZostera marina reveals angiosperm adaptation to the sea.
The Norway spruce genome sequence and conifer genome evolution.
The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla).
Draft genome of the kiwifruitActinidia chinensis.
The collapse of gene complement following whole genome duplication.
Polyploidy: adaptation to the genomic environment.
Abstract Genomic evidence of ancestral whole genome duplication (WGD) and polyploidy is widespread among eukaryotic species, and especially among plants. WGD is thought to provide the raw material for adaptation in the form of duplicated genes, and polyploids are thought to benefit from both physiological and genetic buffering. Comparatively little attention has focused on the genomic challenge of polyploidy, however, although much evidence exists that polyploidy severely perturbs important cellular functions. Here, I review recent progress in the study of the re-establishment of stable meiosis in recently evolved polyploids, focusing on four plant species. This work has yielded an insight into the mechanisms underlying stabilization of genome transmission in polyploids, and is revealing remarkable parallels among diverse taxa. Importantly, these studies also provide a road map for investigating how polyploids respond to the challenge of WGD.
Early genome duplications in conifers and other seed plants.
A new phylogenomic approach reveals that conifer genomes are duplicated despite rare polyploidy among extant species. Polyploidy is a common mode of speciation and evolution in angiosperms (flowering plants). In contrast, there is little evidence to date that whole genome duplication (WGD) has played a significant role in the evolution of their putative extant sister lineage, the gymnosperms. Recent analyses of the spruce genome, the first published conifer genome, failed to detect evidence of WGDs in gene age distributions and attributed many aspects of conifer biology to a lack of WGDs. We present evidence for three ancient genome duplications during the evolution of gymnosperms, based on phylogenomic analyses of transcriptomes from 24 gymnosperms and 3 outgroups. We use a new algorithm to place these WGD events in phylogenetic context: two in the ancestry of major conifer clades (Pinaceae and cupressophyte conifers) and one inWelwitschia(Gnetales). We also confirm that a WGD hypothesized to be restricted to seed plants is indeed not shared with ferns and relatives (monilophytes), a result that was unclear in earlier studies. Contrary to previous genomic research that reported an absence of polyploidy in the ancestry of contemporary gymnosperms, our analyses indicate that polyploidy has contributed to the evolution of conifers and other gymnosperms. As in the flowering plants, the evolution of the large genome sizes of gymnosperms involved both polyploidy and repetitive element activity.
Draft genome of the living fossil Ginkgo biloba.
Abstract BACKGROUND: Ginkgo biloba L. (Ginkgoaceae) is one of the most distinctive plants. It possesses a suite of fascinating characteristics including a large genome, outstanding resistance/tolerance to abiotic and biotic stresses, and dioecious reproduction, making it an ideal model species for biological studies. However, the lack of a high-quality genome sequence has been an impediment to our understanding of its biology and evolution. FINDINGS: The 10.61 Gb genome sequence containing 41,840 annotated genes was assembled in the present study. Repetitive sequences account for 76.58% of the assembled sequence, and long terminal repeat retrotransposons (LTR-RTs) are particularly prevalent. The diversity and abundance of LTR-RTs is due to their gradual accumulation and a remarkable amplification between 16 and 24 million years ago, and they contribute to the long introns and large genome. Whole genome duplication (WGD) may have occurred twice, with an ancient WGD consistent with that shown to occur in other seed plants, and a more recent event specific to ginkgo. Abundant gene clusters from tandem duplication were also evident, and enrichment of expanded gene families indicates a remarkable array of chemical and antibacterial defense pathways. CONCLUSIONS: The ginkgo genome consists mainly of LTR-RTs resulting from ancient gradual accumulation and two WGD events. The multiple defense mechanisms underlying the characteristic resilience of ginkgo are fostered by a remarkable enrichment in ancient duplicated and ginkgo-specific gene clusters. The present study sheds light on sequencing large genomes, and opens an avenue for further genetic and evolutionary research.
Single-molecule sequencing of the desiccation-tolerant grass
VanBuren R. Progress, challenges and the future of crop genomes.
The availability of plant reference genomes has ushered in a new era of crop genomics. More than 100 plant genomes have been sequenced since 2000, 63% of which are crop species. These genome sequences provide insight into architecture, evolution and novel aspects of crop genomes such as the retention of key agronomic traits after whole genome duplication events. Some crops have very large, polyploid, repeat-rich genomes, which require innovative strategies for sequencing, assembly and analysis. Even low quality reference genomes have the potential to improve crop germplasm through genome-wide molecular markers, which decrease expensive phenotyping and breeding cycles. The next stage of plant genomics will require draft genome refinement, building resources for crop wild relatives, resequencing broad diversity panels, and plant ENCODE projects to better understand the complexities of these highly diverse genomes.
Phased diploid genome assembly with single-molecule real-time sequencing.
Abstract While genome assembly projects have been successful in many haploid and inbred species, the assembly of noninbred or rearranged heterozygous genomes remains a major challenge. To address this challenge, we introduce the open-source FALCON and FALCON-Unzip algorithms (https://github.com/PacificBiosciences/FALCON/) to assemble long-read sequencing data into highly accurate, contiguous, and correctly phased diploid genomes. We generate new reference sequences for heterozygous samples including an F1 hybrid of Arabidopsis thaliana, the widely cultivated Vitis vinifera cv. Cabernet Sauvignon, and the coral fungus Clavicorona pyxidata, samples that have challenged short-read assembly approaches. The FALCON-based assemblies are substantially more contiguous and complete than alternate short- or long-read approaches. The phased diploid assembly enabled the study of haplotype structure and heterozygosities between homologous chromosomes, including the identification of widespread heterozygous structural variation within coding sequences.
The pineapple genome and the evolution of CAM photosynthesis.
Abstract Pineapple (Ananas comosus (L.) Merr.) is the most economically valuable crop possessing crassulacean acid metabolism (CAM), a photosynthetic carbon assimilation pathway with high water-use efficiency, and the second most important tropical fruit. We sequenced the genomes of pineapple varieties F153 and MD2 and a wild pineapple relative, Ananas bracteatus accession CB5. The pineapple genome has one fewer ancient whole-genome duplication event than sequenced grass genomes and a conserved karyotype with seven chromosomes from before the 脧聛 duplication event. The pineapple lineage has transitioned from C3 photosynthesis to CAM, with CAM-related genes exhibiting a diel expression pattern in photosynthetic tissues. CAM pathway genes were enriched with cis-regulatory elements associated with the regulation of circadian clock genes, providing the first cis-regulatory link between CAM and circadian clock regulation. Pineapple CAM photosynthesis evolved by the reconfiguration of pathways in C3 plants, through the regulatory neofunctionalization of preexisting genes and not through the acquisition of neofunctionalized genes via whole-genome or tandem gene duplication.
The genome sequence of allopolyploid
The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution.
The domesticated sunflower, Helianthus annuus L., is a global oil crop that has promise for climate change adaptation, because it can maintain stable yields across a wide variety of environmental conditions, including drought. Even greater resilience is achievable through the mining of resistance alleles from compatible wild sunflower relatives, including numerous extremophile species. Here we report a high-quality reference for the sunflower genome (3.6 gigabases), together with extensive transcriptomic data from vegetative and floral organs. The genome mostly consists of highly similar, related sequences and required single-molecule real-time sequencing technologies for successful assembly. Genome analyses enabled the reconstruction of the evolutionary history of the Asterids, further establishing the existence of a whole-genome triplication at the base of the Asterids II clade and a sunflower-specific whole-genome duplication around 29 million years ago. An integrative approach combining quantitative genetics, expression and diversity data permitted development of comprehensive gene networks for two major breeding traits, flowering time and oil metabolism, and revealed new candidate genes in these networks. We found that the genomic architecture of flowering time has been shaped by the most recent whole-genome duplication, which suggests that ancient paralogues can remain in the same regulatory networks for dozens of millions of years. This genome represents a cornerstone for future research programs aiming to exploit genetic diversity to improve biotic and abiotic stress resistance and oil production, while also considering agricultural constraints and human nutritional needs.
A chromosome conformation capture ordered sequence of the barley genome.
Abstract Cereal grasses of the Triticeae tribe have been the major food source in temperate regions since the dawn of agriculture. Their large genomes are characterized by a high content of repetitive elements and large pericentromeric regions that are virtually devoid of meiotic recombination. Here we present a high-quality reference genome assembly for barley (Hordeum vulgare L.). We use chromosome conformation capture mapping to derive the linear order of sequences across the pericentromeric space and to investigate the spatial organization of chromatin in the nucleus at megabase resolution. The composition of genes and repetitive elements differs between distal and proximal regions. Gene family analyses reveal lineage-specific duplications of genes involved in the transport of nutrients to developing seeds and the mobilization of carbohydrates in grains. We demonstrate the importance of the barley reference sequence for breeding by inspecting the genomic partitioning of sequence variation in modern elite germplasm, highlighting regions vulnerable to genetic erosion.
The genome sequences ofArachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut.
Genome sequence of the hot pepper provides insights into the evolution of pungency inCapsicum species.
Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers.
Long-read sequencing uncovers the adaptive topography of a carnivorous plant genome.
Utricularia gibba, the humped bladderwort, is a carnivorous plant that retains a tiny nuclear genome despite at least two rounds of whole genome duplication (WGD) since common ancestry with grapevine and other species. We used a third-generation genome assembly with several complete chromosomes to reconstruct the two most recent lineage-specific...
Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch.
Nature Genetics 49, 904 (2017). doi:10.1038/ng.3862Authors: Jarkko Saloj01rvi, Olli-Pekka Smolander, Kaisa Nieminen, Sitaram Rajaraman, Omid Safronov, Pezhman Safdari, Airi Lammin ...
The genome ofChenopodium quinoa.
The draft genome of Primula veris yields insights into the molecular basis of heterostyly.
Abstract BACKGROUND: The flowering plant Primula veris is a common spring blooming perennial that is widely cultivated throughout Europe. This species is an established model system in the study of the genetics, evolution, and ecology of heterostylous floral polymorphisms. Despite the long history of research focused on this and related species, the continued development of this system has been restricted due the absence of genomic and transcriptomic resources. RESULTS: We present here a de novo draft genome assembly of P. veris covering 301.8聽Mb, or approximately 63% of the estimated 479.22聽Mb genome, with an N50 contig size of 9.5 Kb, an N50 scaffold size of 164 Kb, and containing an estimated 19,507 genes. The results of a RADseq bulk segregant analysis allow for the confident identification of four genome scaffolds that are linked to the P. veris S-locus. RNAseq data from both P. veris and the closely related species P. vulgaris allow for the characterization of 113 candidate heterostyly genes that show significant floral morph-specific differential expression. One candidate gene of particular interest is a duplicated GLOBOSA homolog that may be unique to Primula (PveGLO2), and is completely silenced in L-morph flowers. CONCLUSIONS: The P. veris genome represents the first genome assembled from a heterostylous species, and thus provides an immensely important resource for future studies focused on the evolution and genetic dissection of heterostyly. As the first genome assembled from the Primulaceae, the P. veris genome will also facilitate the expanded application of phylogenomic methods in this diverse family and the eudicots as a whole.
Saccharina genomes provide novel insight into kelp biology
Abstract Seaweeds are essential for marine ecosystems and have immense economic value. Here we present a comprehensive analysis of the draft genome of Saccharina japonica, one of the most economically important seaweeds. The 537-Mb assembled genomic sequence covered 98.5% of the estimated genome, and 18,733 protein-coding genes are predicted and annotated. Gene families related to cell wall synthesis, halogen concentration, development and defence systems were expanded. Functional diversification of the mannuronan C-5-epimerase and haloperoxidase gene families provides insight into the evolutionary adaptation of polysaccharide biosynthesis and iodine antioxidation. Additional sequencing of seven cultivars and nine wild individuals reveal that the genetic diversity within wild populations is greater than among cultivars. All of the cultivars are descendants of a wild S. japonica accession showing limited admixture with S. longissima. This study represents an important advance toward improving yields and economic traits in Saccharina and provides an invaluable resource for plant genome studies.
Analysis of the genome sequence of the medicinal plant
In silico archeogenomics unveils modern plant genome organisation, regulation and evolution
Increasing access to plant genome sequences as well as high resolution gene-based genetic maps have recently offered the opportunity to compare modern genomes and model their evolutionary history from their reconstructed founder ancestors on an unprecedented scale. In silico paleogenomic data have revealed the evolutionary forces that have shaped present-day genomes and allowed us to gain insight into how they are organised and regulated today.
Reconstructing the genome of the most recent common ancestor of flowering plants.
Abstract We describe here the reconstruction of the genome of the most recent common ancestor (MRCA) of modern monocots and eudicots, accounting for 95% of extant angiosperms, with its potential repertoire of 22,899 ancestral genes conserved in present-day crops. The MRCA provides a starting point for deciphering the reticulated evolutionary plasticity between species (rapidly versus slowly evolving lineages), subgenomes (pre- versus post-duplication blocks), genomic compartments (stable versus labile loci), genes (ancestral versus species-specific genes) and functions (gained versus lost ontologies), the key mutational forces driving the success of polyploidy in crops. The estimation of the timing of angiosperm evolution, based on MRCA genes, suggested that this group emerged 214 million years ago during the late Triassic era, before the oldest recorded fossil. Finally, the MRCA constitutes a unique resource for scientists to dissect major agronomic traits in translational genomics studies extending from model species to crops.
Molecular evidence for an ancient duplication of the entire yeast genome.
Gene duplication is an important source of evolutionary novelty. Most duplications are of just a single gene, but Ohno proposed that whole-genome duplication (polyploidy) is an important evolutionary mechanism. Many duplicate genes have been found in Saccharomyces cerevisiae, and these often seem to be phenotypically redundant. Here we show that the arrangement of duplicated genes in the S. cerevisiae genome is consistent with Ohno's hypothesis. We propose a model in which this species is a degenerate tetraploid resulting from a whole-genome duplication that occurred after the divergence of Saccharomyces from Kluyveromyces. Only a small fraction of the genes were subsequently retained in duplicate (most were deleted), and gene order was rearranged by many reciprocal translocations between chromosomes. Protein pairs derived from this duplication event make up 13% of all yeast proteins, and include pairs of transcription factors, protein kinases, myosins, cyclins and pheromones. Tetraploidy may have facilitated the evolution of anaerobic fermentation in Saccharomyces.
Analysis of the genome sequence of the flowering plant
French-Italian Public Consortium for Grapevine Genome Characterization. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla.
ThePhyscomitrella genome reveals evolutionary insights into the conquest of land by plants.
We report the draft genome sequence of the model moss Physcomitrella patens and compare its features with those of flowering plants, from which it is separated by more than 400 million years, and unicellular aquatic algae. This comparison reveals genomic changes concomitant with the evolutionary movement to land, including a general increase in gene family complexity; loss of genes associated with aquatic environments (e.g., flagellar arms); acquisition of genes for tolerating terrestrial stresses (e.g., variation in temperature and water availability); and the development of the auxin and abscisic acid signaling pathways for coordinating multicellular growth and dehydration response. The Physcomitrella genome provides a resource for phylogenetic inferences about gene function and for experimental analysis of plant processes through this plant's unique facility for reverse genetics.
The Selaginella genome identifies genetic changes associated with the evolution of vascular plants.
TheAmborella genome and the evolution of flowering plants.
Amborella trichopoda is strongly supported as the single living species of the sister lineage to all other extant flowering plants, providing a unique reference for inferring the genome content and structure of the most recent common ancestor (MRCA) of living angiosperms. Sequencing the Amborella genome, we identified an ancient genome duplication predating angiosperm diversification, without evidence of subsequent, lineage-specific genome duplications. Comparisons between Amborella and other angiosperms facilitated reconstruction of the ancestral angiosperm gene content and gene order in the MRCA of core eudicots. We identify new gene families, gene duplications, and floral protein-protein interactions that first appeared in the ancestral angiosperm. Transposable elements in Amborella are ancient and highly divergent, with no recent transposon radiations. Population genomic analysis across Amborella's native range in New Caledonia reveals a recent genetic bottleneck and geographic structure with conservation implications.
A phylogenomic assessment of ancient polyploidy and genome evolution across the Poales.
Comparisons of flowering plant genomes reveal multiple rounds of ancient polyploidy characterized by large intragenomic syntenic blocks. Three such whole-genome duplication (WGD) events, designated as rho (蟻), sigma (蟽), and tau (蟿), have been identified in the genomes of cereal grasses. Precise dating of these WGD events is necessary to investigate how they have influenced diversification rates, evolutionary innovations, and genomic characteristics such as the GC profile of protein-coding sequences. The timing of these events has remained uncertain due to the paucity of monocot genome sequence data outside the grass family (Poaceae). Phylogenomic analysis of protein-coding genes from sequenced genomes and transcriptome assemblies from 35 species, including representatives of all families within the Poales, has resolved the timing ofrhoandsigmarelative to speciation events and placedtauprior to divergence of Asparagales and the commelinids but after divergence with eudicots. Examination of gene family phylogenies indicates thatrhooccurred just prior to the diversification of Poaceae andsigmaoccurred before early diversification of Poales lineages but after the Poales-commelinid split. Additional lineage-specific WGD events were identified on the basis of the transcriptome data. Gene families exhibiting high GC content are underrepresented among those with duplicate genes that persisted following these genome duplications. However, genome duplications had little overall influence on lineage-specific changes in the GC content of coding genes. Improved resolution of the timing of WGD events in monocot history provides evidence for the influence of polyploidization on functional evolution and species diversification.
Evaluating and characterizing ancient whole-genome duplications in plants with gene count data.
Whole-genome duplications (WGDs) have helped shape the genomes of land plants, and recent evidence suggests that the genomes of all angiosperms have experienced at least two ancient WGDs. In plants, WGDs often are followed by rapid fractionation, in which many homeologous gene copies are lost. Thus, it can be extremely difficult to identify, let alone characterize, ancient WGDs. In this study, we use a new maximum likelihood estimator to test for evidence of ancient WGDs in land plants and estimate the fraction of new genes copies that are retained following a WGD using gene count data, the number of gene copies in gene families. We identified evidence of many putative ancient WGDs in land plants and found that the genome fractionation rates vary tremendously among ancient WGDs. Analyses of WGDs within Brassicales also indicate that background gene duplication and loss rates vary across land plants, and different gene families have different probabilities of being retained following a WGD. Although our analyses are largely robust to errors in duplication and loss rates and the choice of priors, simulations indicate that this method can have trouble detecting multiple WGDs that occur on the same branch, especially when the gene retention rates for ancient WGDs are very low. They also suggest that we should carefully evaluate evidence for some ancient plant WGD hypotheses.
The value of nonmodel genomes and an example using SynMap within CoGe to dissect the hexaploidy that predates the rosids.
Inference of genome duplications from age distributions revisited.
Abstract Whole-genome duplications (WGDs), thought to facilitate evolutionary innovations and adaptations, have been uncovered in many phylogenetic lineages. WGDs are frequently inferred from duplicate age distributions, where they manifest themselves as peaks against a small-scale duplication background. However, the interpretation of duplicate age distributions is complicated by the use of K(S), the number of synonymous substitutions per synonymous site, as a proxy for the age of paralogs. Two particular concerns are the stochastic nature of synonymous substitutions leading to increasing uncertainty in K(S) with increasing age since duplication and K(S) saturation caused by the inability of evolutionary models to fully correct for the occurrence of multiple substitutions at the same site. K(S) stochasticity is expected to erode the signal of older WGDs, whereas K(S) saturation may lead to artificial peaks in the distribution. Here, we investigate the consequences of these effects on K(S)-based age distributions and WGD inference by simulating the evolution of duplicated sequences according to predefined real age distributions and re-estimating the corresponding K(S) distributions. We show that, although K(S) estimates can be used for WGD inference far beyond the commonly accepted K(S) threshold of 1, K(S) saturation effects can cause artificial peaks at higher ages. Moreover, K(S) stochasticity and saturation may lead to confounded peaks encompassing multiple WGD events and/or saturation artifacts. We argue that K(S) effects need to be properly accounted for when inferring WGDs from age distributions and that the failure to do so could lead to false inferences.
Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement.
Chickpea (Cicer arietinum) is the second most widely grown legume crop after soybean, accounting for a substantial proportion of human dietary nitrogen intake and playing a crucial role in food security in developing countries. We report the 鈭738-Mb draft whole genome shotgun sequence of CDC Frontier, a kabuli chickpea variety, which contains an estimated 28,269 genes. Resequencing and analysis of 90 cultivated and wild genotypes from ten countries identifies targets of both breeding-associated genetic sweeps and breeding-associated balancing selection. Candidate genes for disease resistance and agronomic traits are highlighted, including traits that distinguish the two main market classes of cultivated chickpea--desi and kabuli. These data comprise a resource for chickpea improvement through molecular breeding and provide insights into both genome diversity and domestication.
Genome sequence of the palaeopolyploid soybean.
Genome sequence of the palaeopolyploid soybeanNature 465, 120 (2010). doi:10.1038/nature08957Authors: Jeremy Schmutz, Steven B. Cannon, Jessica Schlueter, Jianxin Ma, Therese Mitros, Willi ...
The genome sequence of the orchidPhalaenopsis equestris.
Analyses of transcriptome sequences reveal multiple ancient large-scale duplication events in the ancestor of Sphagnopsida (Bryophyta).
Abstract The goal of this research was to investigate whether there has been a whole-genome duplication (WGD) in the ancestry of Sphagnum (peatmoss) or the class Sphagnopsida, and to determine if the timing of any such duplication(s) and patterns of paralog retention could help explain the rapid radiation and current ecological dominance of peatmosses. RNA sequencing (RNA-seq) data were generated for nine taxa in Sphagnopsida (Bryophyta). Analyses of frequency plots for synonymous substitutions per synonymous site (K s ) between paralogous gene pairs and reconciliation of 578 gene trees were conducted to assess evidence of large-scale or genome-wide duplication events in each transcriptome. Both K s frequency plots and gene tree-based analyses indicate multiple duplication events in the history of the Sphagnopsida. The most recent WGD event predates divergence of Sphagnum from the two other genera of Sphagnopsida. Duplicate retention is highly variable across species, which might be best explained by local adaptation. Our analyses indicate that the last WGD could have been an important factor underlying the diversification of peatmosses and facilitated their rise to ecological dominance in peatlands. The timing of the duplication events and their significance in the evolutionary history of peat mosses are discussed. 漏 2016 The Authors. New Phytologist 漏 2016 New Phytologist Trust.
Large-Scale gene relocations following an ancient genome triplication associated with the diversification of core eudicots.
Abstract Different modes of gene duplication including whole-genome duplication (WGD), and tandem, proximal and dispersed duplications are widespread in angiosperm genomes. Small-scale, stochastic gene relocations and transposed gene duplications are widely accepted to be the primary mechanisms for the creation of dispersed duplicates. However, here we show that most surviving ancient dispersed duplicates in core eudicots originated from large-scale gene relocations within a narrow window of time following a genome triplication (γ) event that occurred in the stem lineage of core eudicots. We name these surviving ancient dispersed duplicates as relocated γ duplicates. In Arabidopsis thaliana, relocated γ, WGD and single-gene duplicates have distinct features with regard to gene functions, essentiality, and protein interactions. Relative to γ duplicates, relocated γ duplicates have higher non-synonymous substitution rates, but comparable levels of expression and regulation divergence. Thus, relocated γ duplicates should be distinguished from WGD and single-gene duplicates for evolutionary investigations. Our results suggest large-scale gene relocations following the γ event were associated with the diversification of core eudicots.
Whole genome duplications in plants: an overview fromArabidopsis.
Abstract Polyploidy is a common event in plants that involves the acquisition of more than two complete sets of chromosomes. Allopolyploidy originates from interspecies hybrids while autopolyploidy originates from intraspecies whole genome duplication (WGD) events. In spite of inconveniences derived from chromosomic rearrangement during polyploidization, natural plant polyploids species often exhibit improved growth vigour and adaptation to adverse environments, conferring evolutionary advantages. These advantages have also been incorporated into crop breeding programmes. Many tetraploid crops show increased stress tolerance, although the molecular mechanisms underlying these different adaptation abilities are poorly known. Understanding the physiological, cellular, and molecular mechanisms coupled to WGD, in both allo- and autopolyploidy, is a major challenge. Over the last few years, several studies, many of them in Arabidopsis, are shedding light on the basis of genetic, genomic, and epigenomic changes linked to WGD. In this review we summarize and discuss the latest advances made in Arabidopsis polyploidy, but also in other agronomic plant species. 漏 The Author 2015. Published by Oxford University Press on behalf of the Society for Experimental Biology. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea.
Abstract BACKGROUND: Brassica oleracea is a valuable vegetable species that has contributed to human health and nutrition for hundreds of years and comprises multiple distinct cultivar groups with diverse morphological and phytochemical attributes. In addition to this phenotypic wealth, B. oleracea offers unique insights into polyploid evolution, as it results from multiple ancestral polyploidy events and a final Brassiceae-specific triplication event. Further, B. oleracea represents one of the diploid genomes that formed the economically important allopolyploid oilseed, Brassica napus. A deeper understanding of B. oleracea genome architecture provides a foundation for crop improvement strategies throughout the Brassica genus. RESULTS: We generate an assembly representing 75% of the predicted B. oleracea genome using a hybrid Illumina/Roche 454 approach. Two dense genetic maps are generated to anchor almost 92% of the assembled scaffolds to nine pseudo-chromosomes. Over 50,000 genes are annotated and 40% of the genome predicted to be repetitive, thus contributing to the increased genome size of B. oleracea compared to its close relative B. rapa. A snapshot of both the leaf transcriptome and methylome allows comparisons to be made across the triplicated sub-genomes, which resulted from the most recent Brassiceae-specific polyploidy event. CONCLUSIONS: Differential expression of the triplicated syntelogs and cytosine methylation levels across the sub-genomes suggest residual marks of the genome dominance that led to the current genome architecture. Although cytosine methylation does not correlate with individual gene dominance, the independent methylation patterns of triplicated copies suggest epigenetic mechanisms play a role in the functional diversification of duplicate genes.
The genome of the mesopolyploid crop species
Whole genome duplication in coast redwood (Sequoia sempervirens) and its implications for explaining the rarity of polyploidy in conifers.
Abstract Polyploidy is common and an important evolutionary factor in most land plant lineages, but it is rare in gymnosperms. Coast redwood (Sequoia sempervirens) is one of just two polyploid conifer species and the only hexaploid. Evidence from fossil guard cell size suggests that polyploidy in Sequoia dates to the Eocene. Numerous hypotheses about the mechanism of polyploidy and parental genome donors have been proposed, based primarily on morphological and cytological data, but it remains unclear how Sequoia became polyploid and why this lineage overcame an apparent gymnosperm barrier to whole-genome duplication (WGD). We sequenced transcriptomes and used phylogenetic inference, Bayesian concordance analysis and paralog age distributions to resolve relationships among gene copies in hexaploid coast redwood and close relatives. Our data show that hexaploidy in coast redwood is best explained by autopolyploidy or, if there was allopolyploidy, it happened within the Californian redwood clade. We found that duplicate genes have more similar sequences than expected, given the age of the inferred polyploidization. Conflict between molecular and fossil estimates of WGD can be explained if diploidization occurred very slowly following polyploidization. We extrapolate from this to suggest that the rarity of polyploidy in gymnosperms may be due to slow diploidization in this clade. 脗漏 2016 The Authors. New Phytologist 脗漏 2016 New Phytologist Trust.
The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions.
Watermelon, Citrullus lanatus, is an important cucurbit crop grown throughout the world. Here we report a high-quality draft genome sequence of the east Asia watermelon cultivar 97103 (2n = 2脙鈥 = 22) containing 23,440 predicted protein-coding genes. Comparative genomics analysis provided an evolutionary scenario for the origin of the 11 watermelon chromosomes derived from a 7-chromosome paleohexaploid eudicot ancestor. Resequencing of 20 watermelon accessions representing three different C. lanatus subspecies produced numerous haplotypes and identified the extent of genetic diversity and population structure of watermelon germplasm. Genomic regions that were preferentially selected during domestication were identified. Many disease-resistance genes were also found to be lost during domestication. In addition, integrative genomic and transcriptomic analyses yielded important insights into aspects of phloem-based vascular signaling in common between watermelon and cucumber and identified genes crucial to valuable fruit-quality traits, including sugar accumulation and citrulline metabolism.
Genome sequencing and analysis of the model grassBrachypodium distachyon.
Abstract Top of page Abstract Literature cited Ribosomal cistrons for 28S and 18S RNA have been located in the mitotic chromosomes of several species at the nucleolus organizer region, but when amplification occurs at meiosis the number of chromomeres per site and the number of sites involved in this process have not been determined. In Acheta domesticus where amplification occurs for these genes every chromosome of the complement has been identified at pachytene by its size and characteristic chromomere markers both in oocytes and spermatocytes. In oocytes there are 5 amplification sites. Of these, two are major chromomeres, or DNA bodies, which are localized in the autosomes Nos. 6 and 11. Since these major chromomeres are absent from the chromosomes of spermatocytes, a comparison of the chromosomes of the two tissues at pachytene makes it possible to determine the size of the amplicons (the regions of amplification) in this species. The amplicon of chromosome 6 consists of 2 chromomere pairs and that of chromosome 11 of one chromomere pair. The total chromomere number of the autosomes is 185.3 in the oocytes and 178.4 in the spermatocytes. The X chromosome is an exception since it has 41.7 chromomeres in the oocytes, whereas in the spermatocytes it has none. The significance of these data for the understanding of gene amplification is discussed.
The presumed ginkgophyteUmaltolepis has seed-bearing structures resembling those of Peltaspermales and Umkomasiales.
Abstract The origins of the five groups of living seed plants, including the single relictual species Ginkgo biloba , are poorly understood, in large part because of very imperfect knowledge of extinct seed plant diversity. Here we describe well-preserved material from the Early Cretaceous of Mongolia of the previously enigmatic Mesozoic seed plant reproductive structure Umaltolepis , which has been presumed to be a ginkgophyte. Abundant new material shows that Umaltolepis is a seed-bearing cupule that was borne on a stalk at the tip of a short shoot. Each cupule is umbrella-like with a central column that bears a thick, resinous, four-lobed outer covering, which opens from below. Four, pendulous, winged seeds are attached to the upper part of the column and are enclosed by the cupule. Evidence from morphology, anatomy, and field association suggests that the short shoots bore simple, elongate Pseudotorellia leaves that have similar venation and resin ducts to leaves of living Ginkgo Umaltolepis seed-bearing structures are very different from those of Ginkgo but very similar to fossils described previously as Vladimaria. Umaltolepis and Vladimaria do not closely resemble the seed-bearing structures of any living or extinct plant, but are comparable in some respects to those of certain Peltaspermales and Umkomasiales (corystosperms). Vegetative similarities of the Umaltolepis plant to Ginkgo , and reproductive similarities to extinct peltasperms and corystosperms, support previous ideas that Ginkgo may be the last survivor of a once highly diverse group of extinct plants, several of which exhibited various degrees of ovule enclosure.
Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies.
Background The size and complexity of conifer genomes has, until now, prevented full genome sequencing and assembly. The large research community and economic importance of loblolly pine, Pinus taeda L., made it an early candidate for reference sequence determination. Results We develop a novel strategy to sequence the genome of loblolly pine that combines unique aspects of pine reproductive biology and genome assembly methodology. We use a whole genome shotgun approach relying primarily on next generation sequence generated from a single haploid seed megagametophyte from a loblolly pine tree, 20-1010, that has been used in industrial forest tree breeding. The resulting sequence and assembly was used to generate a draft genome spanning 23.2 Gbp and containing 20.1 Gbp with an N50 scaffold size of 66.9 kbp, making it a significant improvement over available conifer genomes. The long scaffold lengths allow the annotation of 50,172 gene models with intron lengths averaging over 2.7 kbp and sometimes exceeding 100 kbp in length. Analysis of orthologous gene sets identifies gene families that may be unique to conifers. We further characterize and expand the existing repeat library based on the de novo analysis of the repetitive content, estimated to encompass 82% of the genome. Conclusions In addition to its value as a resource for researchers and breeders, the loblolly pine genome sequence and assembly reported here demonstrates a novel approach to sequencing the large and complex genomes of this important group of plants that can now be widely applied.
Genome mapping in plant comparative genomics.
Recently, advances in genome-mapping technology have made this resource more widely available. Improved optics, advanced molecular biology, informatics tools, and creative innovations have been combined to create relatively low-cost mapping tools.
Ancient WGD events as drivers of key innovations in angiosperms.
Polyploidy, or whole-genome duplication (WGD), is a ubiquitous feature of plant genomes, contributing to variation in both genome size and gene content. Although polyploidy has occurred in all major clades of land plants, it is most frequent in angiosperms. Following a WGD in the common ancestor of all extant angiosperms, a complex pattern of both ancient and recent polyploidy is evident across angiosperm phylogeny. In several cases, ancient WGDs are associated with increased rates of species diversification. For example, a WGD in the common ancestor of Asteraceae, the largest family of angiosperms with 6525000 species, is statistically linked to a shift in species diversification; several other old WGDs are followed by increased diversification after a ‘lag’ of up to three nodes. WGD may thus lead to a genomic combination that generates evolutionary novelty and may serve as a catalyst for diversification. In this paper, we explore possible links between WGD, the origin of novelty, and key innovations and propose a research path forward.
Evolution of gene duplication in plants.
Ancient duplication events and a high rate of retention of extant pairs of duplicate genes have contributed to an abundance of duplicate genes in plant genomes. These duplicates have contributed to the evolution of novel functions, such as the production of floral structures, induction of disease resistance, and adaptation to stress. Additionally, recent whole-genome duplications that have occurred in the lineages of several domesticated crop species, including wheat (Triticum aestivum), cotton (Gossypium hirsutum), and soybean (Glycine max), have contributed to important agronomic traits, such as grain quality, fruit shape, and flowering time. Therefore, understanding the mechanisms and impacts of gene duplication will be important to future studies of plants in general and of agronomically important crops in particular. In this review, we survey the current knowledge about gene duplication, including gene duplication mechanisms, the potential fates of duplicate genes, models explaining duplicate gene retention, the properties that distinguish duplicate from singleton genes, and the evolutionary impact of gene duplication.
Evaluating the role of genome downsizing and size thresholds from genome size distributions in angiosperms.
Abstract Premise of the study: Whole-genome duplications (WGDs) can rapidly increase genome size in angiosperms. Yet their mean genome size is not correlated with ploidy. We compared three hypotheses to explain the constancy of genome size means across ploidies. The genome downsizing hypothesis suggests that genome size will decrease by a given percentage after a WGD. The genome size threshold hypothesis assumes that taxa with large genomes or large monoploid numbers will fail to undergo or survive WGDs. Finally, the genome downsizing and threshold hypothesis suggests that both genome downsizing and thresholds affect the relationship between genome size means and ploidy. Methods: We performed nonparametric bootstrap simulations to compare observed angiosperm genome size means among species or genera against simulated genome sizes under the three different hypotheses. We evaluated the hypotheses using a decision theory approach and estimated the expected percentage of genome downsizing. Key results: The threshold hypothesis improves the approximations between mean genome size and simulated genome size. At the species level, the genome downsizing with thresholds hypothesis best explains the genome size means with a 15% genome downsizing percentage. In the genus level simulations, the monoploid number threshold hypothesis best explains the data. Conclusions: Thresholds of genome size and monoploid number added to genome downsizing at species level simulations explain the observed means of angiosperm genome sizes, and monoploid number is important for determining the genome size mean at the genus level.
The collapse of gene complement following whole genome duplication.
Data access for the 1, 000 Plants (1KP) project.
Abstract The 1,000 plants (1KP) project is an international multi-disciplinary consortium that has generated transcriptome data from over 1,000 plant species, with exemplars for all of the major lineages across the Viridiplantae (green plants) clade. Here, we describe how to access the data used in a phylogenomics analysis of the first 85 species, and how to visualize our gene and species trees. Users can develop computational pipelines to analyse these data, in conjunction with data of their own that they can upload. Computationally estimated protein-protein interactions and biochemical pathways can be visualized at another site. Finally, we comment on our future plans and how they fit within this scalable system for the dissemination, visualization, and analysis of large multi-species data sets.



{kind=link}
{kind=link}
{kind=link}
{kind=link}