
遗传 ›› 2025, Vol. 47 ›› Issue (3): 329-341.doi: 10.16288/j.yczz.24-228
关静1,2( ), 吴萧男1,2, 李进1,2, 谌国会1,2, 王洪阳1,2, 王秋菊1,2()
), 吴萧男1,2, 李进1,2, 谌国会1,2, 王洪阳1,2, 王秋菊1,2()
收稿日期:2024-09-14
修回日期:2024-12-23
出版日期:2025-03-20
发布日期:2025-01-06
通讯作者:
关静,博士,主任医师、副教授,研究方向:聋病遗传学诊断与遗传咨询。E-mail: ggy3u@126.com;基金资助:
Jing Guan1,2(), XiaonanWu 1,2, Jin Li1,2, Guohui Chen1,2, Hongyang Wang1,2, Qiuju Wang1,2()
Received:2024-09-14
Revised:2024-12-23
Published:2025-03-20
Online:2025-01-06
Supported by:摘要:
新发突变(de novo mutation, DNM)是导致散发耳聋的重要遗传因素,也是复杂耳聋综合征发病的重要致病原因。为了分析散发耳聋DNM遗传学特征及其致病因素,本文以2015年10月~2023年10月纳入“中国聋病基因组计划”的410个散发耳聋核心家系为对象,对收集到的先证者及其父母的临床信息进行了回顾性分析,同时通过靶向捕获高通量测序、线粒体基因组及全基因组拷贝数变异检测,进行了“父+母+先证者”核心家系遗传学比对分析,应用同源等位基因检测方法来计算DNM先证者核心家系成员之间关系系数。结果发现,这些散发耳聋核心家系中有7.3%(30例)先证者携带17种常染色体显性基因新发SNV、Indel和1种新发拷贝数变异,涵盖所有DNM类型,其中WFS1c.2051C>T、ATP1A3 c.2452G>A、ACTG1 c.94C>T是散发耳聋中常见DNM,基因型C>T变异占比最高(34.6%);临床特征分析也显示有56.7%(17/30)先证者为非综合征性耳聋,但其中有半数以上(52.9%,9/17)携带明确与“综合征性耳聋”相关的致病基因型,可能存在暂时性“拟”非综合征性耳聋表型特征。本组30例先证者父母的平均生育年龄分别为29.4岁和28.3岁,其中父亲或母亲生育年龄≥35岁的各占13.3%;同时在遗传咨询的先证者家庭结构中,63.3%为独生子女家庭且有明确再生育意愿,16.7%先证者父母为孕育“二孩”产前遗传咨询。遗传咨询时,需要以“父+母+先证者”核心家系为单位进行检测,以便分析DNM在聋病发生中的遗传贡献度;由于DNM发生与父母生育年龄的增加存在一定相关性,因此对于已生育DNM散发耳聋患者家庭,还需要采集听力健康父母的生育年龄及孕产史等临床信息,当这些家庭再生育时建议受孕后进行已明确DNM致病变异的产前诊断并重视妊娠结局。
关静, 吴萧男, 李进, 谌国会, 王洪阳, 王秋菊. 基于散发耳聋核心家系的基因新发突变(DNM)特征分析及遗传咨询策略[J]. 遗传, 2025, 47(3): 329-341.
Jing Guan, XiaonanWu , Jin Li, Guohui Chen, Hongyang Wang, Qiuju Wang. Interpretation of de novo mutations (DNM) and genetic counseling for sporadic hearing loss based on family trio-based sequencing[J]. Hereditas(Beijing), 2025, 47(3): 329-341.
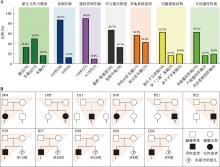
图1
本组30例DNM患者特征及其多子女核心家系结构特征 A:30例DNM患者的临床特征;B:本组多子女核心家系的结构特征。"
表1
30例DNM耳聋患者的致病基因型及临床特征"
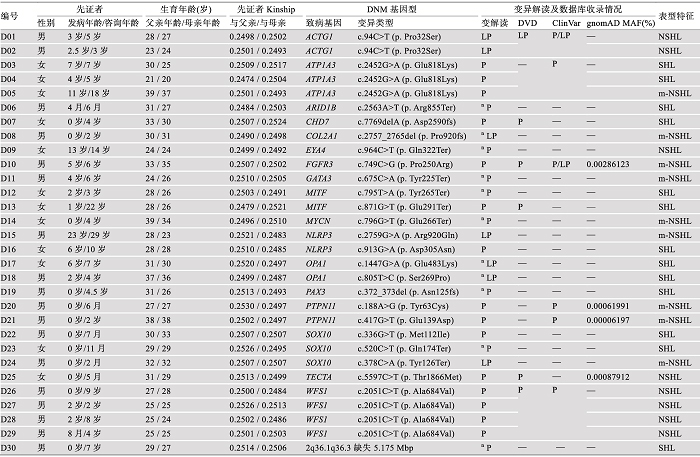 |
表2
本组致病基因DNMs患者表型与既往文献报道表型特征总结"
| 基因 | OMIM 遗传模式 | 表型特征(发病年龄、HL特征、是否伴有其他系统) | |
|---|---|---|---|
| 本组患者 | 文献报道 | ||
| ACTG1 | AD | 2例患者,2~3岁发病,呈中度或极重度、平坦型HL,不伴其他系统症状 | 常染色体显性遗传DFNA20/26型耳聋,发病年龄13.2±4.6岁,呈双耳斜坡型、感音神经性HL,随年龄进行性加重,逐渐累及全频、呈极重度HL[ |
| ATP1A3 | AD | 3例患者,4~11岁发病,呈上坡型/低中频型或平坦型、中度或重度HL,言语识别率差,诊断听神经病。其中1例患者内听道MRI显示双侧蜗神经纤细,2例患者伴视神经萎缩,不伴其他神经系统症状 | CAPOS综合征(小脑性共济失调、反射消失、高弓足、视神经萎缩和感音神经性听力损失),6月龄~3岁发病;其中HL和视神经萎缩常于3岁时出现,HL呈轻度或中重度上坡型;所有症状进展缓慢[ |
| ARID1B | AD | 1例患者,4月龄发病,呈极重度、平坦型HL;伴发育迟缓、轻度腭裂、隐睾 | Coffin-Siris综合征(发育障碍、严重言语障碍、小头畸形、面部特征粗糙、第5手指/脚趾指甲发育不全或缺如),无明确听力学描述[ |
| CHD7 | AD | 1例患者,先天性,呈重度、平坦型HL,伴心血管系统主动脉异常,诊断为非典型CHARGE综合征 | CHARGE综合征(眼盲症;心异常;后鼻孔闭锁;智力和躯体发育迟缓;小阴茎;耳部结构发育异常和/或耳聋)[ |
| COL2A1 | AD | 1例患者,先天性,呈中度、平坦型HL,不伴其他系统症状 | Stickler综合征(严重视力、听力和关节疾病,婴儿期或儿童期发病;眼睛突出、鼻孔前倾、下巴后缩、腭裂等面部特征,高度近视、青光眼、听力下降、骨骼关节异常)[ |
| EYA4 | AD | 1例患者,13岁发病,呈中度、中高频下降型HL;不伴其他系统症状 | 常染色体显性遗传DFNA10型耳聋,呈双侧对称、迟发性、渐进性下降。已报道发病年龄4~50岁,发病初期常累及中频听力,呈谷型或平坦型,约50岁以后进展为重度-极重度耳聋。也有报道成年伴有扩张型心肌病的个案[ |
| FGFR3 | AD | 1例患者,5岁发病,呈轻度、U型HL,言语发育良好;不伴其他系统症状 | 已报道多种不同表型,包括:颅缝早闭,CATSHL综合征(屈指畸形、高个子,脊柱侧凸、听力损失,HL先天或儿童期发病、程度从轻度到重度均有报道),软骨发育不全,脂肪代谢异常,Muenke综合征,Crouzon综合征伴黑棘皮病,严重软骨发育不全伴发育迟缓和黑棘皮病,LADD综合征(泪-耳-牙-指综合征)[ |
| GATA3 | AD | 1例患者,4岁发病,呈中度、斜坡型HL | HDR综合征(甲状旁腺功能减退、感音神经性耳聋、肾发育不良);HL是最常见的特点,最早可于出生时发病,程度轻重不等[ |
| MITF | AD | 2例患者,1~2岁发病,呈重度-极重度、平坦型HL,伴虹膜异色,诊断为Waardenburg综合征 | Waardenburg综合征II型(感音神经性耳聋、虹膜异色、额白发/早白发,不伴内眦间距异常)[ |
| MYCN | AD | 1例患者,先天性,呈极重度、平坦型HL | Feingold综合征(小头畸形、肢体畸形、食管和十二指肠闭锁以及学习障碍/智力迟钝等症状);巨脑-多指综合征[ |
| NLRP3 | AD | 2例患者,6~23岁发病,呈中度、斜坡型HL;1例成年患者无其他系统症状;1例儿童患者伴有周期性发热、荨麻疹、结膜炎,诊断为CINCA综合征 | 常染色体显性遗传DFNA34型耳聋,伴或不伴炎性症状(荨麻疹、周期性发热、结膜炎、关节炎等);CINCA综合征(慢性婴儿神经皮肤关节综合征),新生儿发病是出现反复发热、皮疹,随年龄逐渐出现HL、关节炎[ |
| OPA1 | AD | 2例患者,2~6岁发病,呈中度、上坡型/低中频型HL,听神经病伴视神经萎缩 | 听神经病、视神经萎缩[ |
| PAX3 | AD | 1例患者,先天性,呈极重度、平坦型HL,伴虹膜异色、内眦间距增宽,诊断为Waardenburg综合征 | 色素-听力综合征:先天性感音神经性聋、虹膜色素异常、毛发皮肤色素异常等[ |
| PTPN11 | AD | 2例患者,先天性,呈极重度、平坦型HL,不伴其他系统症状 | Noonan综合征;LEOPARD综合征[ |
| SOX10 | AD | 3例患者,先天性极重度、平坦型HL; 2例伴单侧虹膜异色,诊断为Waardenburg综合征;1例不伴其他系统症状 | Waardenburg综合征2型和Waardenburg综合征4型,典型表型:额白发、虹膜色素缺失、四肢末端皮肤变白、先天性极重度感音神经性耳聋[ |
| TECTA | AD | 1例患者,先天性,呈重度、平坦型HL,不伴其他系统症状 | DFNA8/12:语前聋,中频下降型[ |
| WFS1 | AD | 4例患者,≤2岁发病,呈极重度、平坦型HL,不伴其他系统症状 | DFNA6/14/38:语前聋,低频型、呈进行性;WLS综合征:进行听力下降伴视神经萎缩或葡萄糖调节耐量受损[ |
| 2q36.1q36.3 缺失 | — | 1例患者,先天性,呈极重度、平坦型HL,伴颅面畸形(眼距宽、鼻梁扁平)、严重龋齿,生长发育迟缓、拇指屈曲、断掌、手掌向尺侧偏斜 | 2q36缺失综合征,表型异质性较大,相关表型特征包括智力及运动发育迟缓、内眦距离增宽、手或脚趾畸形、脊柱侧凸或脊柱裂、听力损失、牙齿发育异常[ |

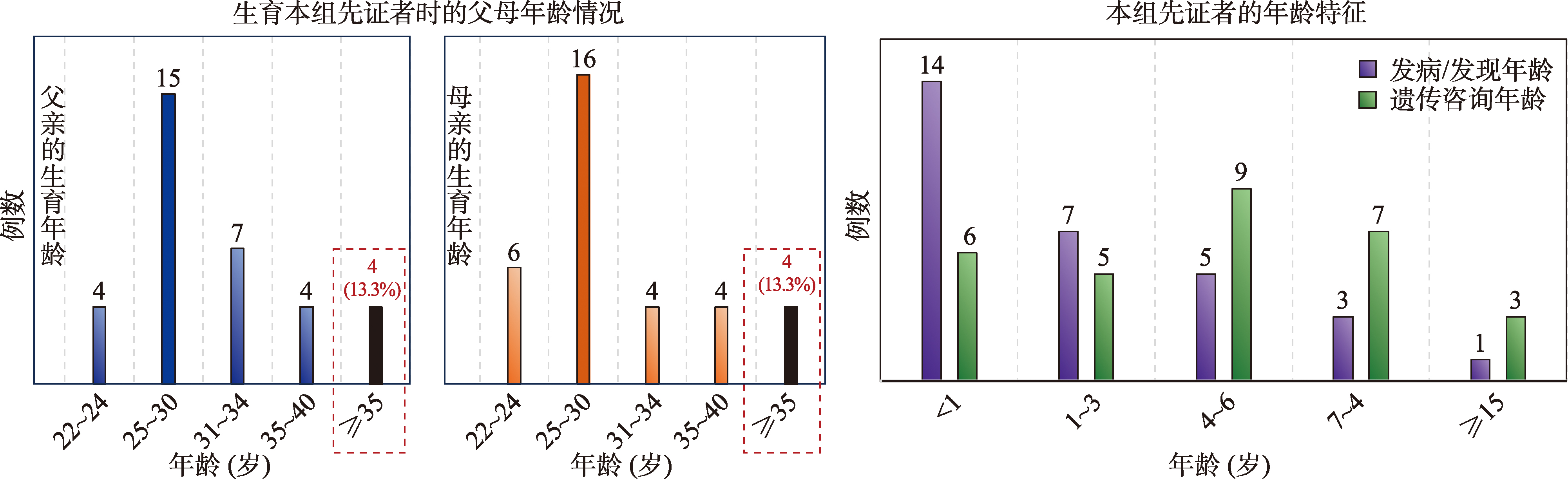
图2
父母生育年龄和先证者年龄特征 红框所示为高龄组父母例数和所占比例。"
| [1] |
Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, UK10K Consortium, Hurles ME. Timing, rates and spectra of human germline mutation. Nat Genet, 2016, 48(2): 126-133.
doi: 10.1038/ng.3469 pmid: 26656846 |
| [2] |
Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol, 2016, 17(1): 241.
pmid: 27894357 |
| [3] | Ambalavanan A, Girard SL, Ahn K, Zhou SR, Dionne- Laporte A, Spiegelman D, Bourassa CV, Gauthier J, Hamdan FF, Xiong L, Dion PA, Joober R, Rapoport J, Rouleau GA. De novo variants in sporadic cases of childhood onset schizophrenia. Eur J Hum Genet, 2016, 24(6): 944-948. |
| [4] | Guan J, Li J, Chen GH, Shi T, Lan L, Wu XN, Zhao C, Wang DY, Wang HY, Wang QJ. Family trio-based sequencing in 404 sporadic bilateral hearing loss patients discovers recessive and De novo genetic variants in multiple ways. Eur J Med Genet, 2021, 64(10): 104311. |
| [5] |
Baux D, Vaché C, Blanchet C, Willems M, Baudoin C, Moclyn M, Faugère V, Touraine R, Isidor B, Dupin- Deguine D, Nizon M, Vincent M, Mercier S, Calais C, García-García G, Azher Z, Lambert L, Perdomo-Trujillo Y, Giuliano F, Claustres M, Koenig M, Mondain M, Roux AF. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci Rep, 2017, 7(1): 16783.
doi: 10.1038/s41598-017-16846-9 pmid: 29196752 |
| [6] |
Cabanillas R, Diñeiro M, Cifuentes GA, Castillo D, Pruneda PC, Álvarez R, Sánchez-Durán N, Capín R, Plasencia A, Viejo-Díaz M, García-González N, Hernando I, Llorente JL, Repáraz-Andrade A, Torreira-Banzas C, Rosell J, Govea N, Gómez-Martínez JR, Núñez-Batalla F, Garrote JA, Mazón-Gutiérrez Á, Costales M, Isidoro- García M, García-Berrocal B, Ordóñez GR, Cadiñanos J. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med Genomics, 2018, 11(1): 58.
doi: 10.1186/s12920-018-0375-5 pmid: 29986705 |
| [7] |
Klimara MJ, Nishimura C, Wang DH, Kolbe DL, Schaefer AM, Walls WD, Frees KL, Smith RJH, Azaiez H. De novo variants are a common cause of genetic hearing loss. Genet Med, 2022, 24(12): 2555-2567.
doi: 10.1016/j.gim.2022.08.028 pmid: 36194208 |
| [8] | Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WSW, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K. Rate of de novo mutations and the importance of father's age to disease risk. Nature, 2012, 488(7412): 471-475. |
| [9] | Guan J, He L, Yang SM, Wang QJ. Expert consensus on genetic counseling for hearing loss. Chin J Otol, 2022, 20(2): 222-226. |
| 关静, 贺林, 杨仕明, 王秋菊. 聋病遗传咨询专家共识. 中华耳科学杂志, 2022, 20(2): 222-226. | |
| [10] |
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015, 17(5): 405-424.
doi: 10.1038/gim.2015.30 pmid: 25741868 |
| [11] |
Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, Shen J, Chapin A, Boczek NJ, Schimmenti LA, Murry JB, Hasadsri L, Nara K, Kenna M, Booth KT, Azaiez H, Griffith A, Avraham KB, Kremer H, Rehm HL, Amr SS, Abou Tayoun AN, ClinGen Hearing Loss Clinical Domain Working Group. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat, 2018, 39(11): 1593-1613.
doi: 10.1002/humu.23630 pmid: 30311386 |
| [12] | Patel MJ, DiStefano MT, Oza AM, Hughes MY, Wilcox EH, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, Shen J, Chapin A, Boczek NJ, Schimmenti LA, Nara K, Kenna M, Azaiez H, Booth KT, Avraham KB, Kremer H, Griffith AJ, Rehm HL, Amr SS, Tayoun ANA, ClinGen Hearing Loss Clinical Domain Working Group. Disease- specific ACMG/AMP guidelines improve sequence variant interpretation for hearing loss. Genet Med, 2021, 23(11): 2208-2212. |
| [13] |
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, Pineda-Alvarez D, Aradhya S, Martin CL. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med, 2020, 22(2): 245-257.
doi: 10.1038/s41436-019-0686-8 pmid: 31690835 |
| [14] | Wang WJ, Li J, Lan L, Xie LY, Xiong F, Guan J, Wang HY, Wang QJ. Auditory neuropathy as the initial phenotype for patients with ATP1A3 c.2452 G>A: Genotype-phenotype study and CI management. Front Cell Dev Biol, 2021, 9: 749484. |
| [15] | Zhang QJ, Lan L, Xie LY, Zhao C, Guan J, Wang QJ. Identification of a novel mutation of SOX10 gene and analysis of the phenotype. Chin J Otorhinolaryngol Head Neck Surg, 2020, 55(11): 1050-1056. |
| 张秋静, 兰兰, 谢林怡, 赵翠, 关静, 王秋菊. SOX10基因新突变的鉴定及其临床表型特征分析. 中华耳鼻咽喉头颈外科杂志, 2020, 55(11): 1050-1056. | |
| [16] |
Hodgkinson A, Eyre-Walker A. Variation in the mutation rate across mammalian genomes. Nat Rev Genet, 2011, 12(11): 756-766.
doi: 10.1038/nrg3098 pmid: 21969038 |
| [17] |
Campbell CD, Eichler EE. Properties and rates of germline mutations in humans. Trends Genet, 2013, 29(10): 575-584.
doi: 10.1016/j.tig.2013.04.005 pmid: 23684843 |
| [18] | Guan J, Wu XN, Zhang J, Li J, Wang HY, Wang QJ. Global research landscape on the contribution of de novo mutations to human genetic diseases over the past 20 years: bibliometric analysis. J Neurogenet, 2024, 38(1): 9-18. |
| [19] | Li C, Chen RY, Fan X, Luo JS, Qian JL, Wang J, Xie BB, Shen YP, Chen SK. EPHA4 haploinsufficiency is responsible for the short stature of a patient with 2q35-q36.2 deletion and Waardenburg syndrome. BMC Med Genet, 2015, 16: 23. |
| [20] |
Song J, Feng Y, Acke FR, Coucke P, Vleminckx K, Dhooge IJ. Hearing loss in Waardenburg syndrome: a systematic review. Clin Genet, 2016, 89(4): 416-425.
doi: 10.1111/cge.12631 pmid: 26100139 |
| [21] | Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature, 2017, 549(7673): 519-522. |
| [22] | Ma KX, Zhang WY, Wang X. Influence of paternal age on progeny prognosis. Chin J Obstet Gynecol, 2021, 56(3): 222-225. |
| 马可心, 张为远, 王欣. 父亲生育年龄对子代预后的影响. 中华妇产科杂志, 2021, 56(3): 222-225. | |
| [23] | Zhu WJ. Opportunities and challenges on fertility for aging men. Chin J Reprod Contracep, 2019, 39(6): 433-435. |
| 朱伟杰. 高龄男性生育研究的机遇与挑战. 中华生殖与避孕杂志, 2019, 39(6): 433-435. | |
| [24] | Lu BY, Han B, Hu HW, Long W, Wang L, Cai ZM, Wang HY, Yu B. Changes in maternal age and its influences on maternal and neonatal complications under the two-child policy. Chin J Perinat Med, 2019, 22(3): 157-163. |
| 陆蓓亦, 韩波, 胡慧文, 龙伟, 王丽, 蔡正茂, 王慧艳, 虞斌. 新生育政策下孕妇年龄的变化及对母婴并发症的影响. 中华围产医学杂志, 2019, 22(3):157-163. | |
| [25] | Mao YY, Hu H, Chen DY, Du YS, Fang YH, Wang SM, Li M, Zhou WJ. Association between parental characteristics during peri-conceptional period and risk of autism spectrum disorders in children. Chin J Reprod Contracep, 2022, 42(4): 372-378. |
| 毛燕燕, 胡宏, 陈东燕, 杜亚松, 房宇航, 王尚明, 李敏, 周维谨. 父母亲生育年龄等围孕期因素与儿童孤独症谱系障碍的关联性. 中华生殖与避孕杂志, 2022, 42(4): 372-378. | |
| [26] |
Acuna-Hidalgo R, Bo T, Kwint MP, van de Vorst M, Pinelli M, Veltman JA, Hoischen A, Vissers LELM, Gilissen C. Post-zygotic point mutations are an underrecognized source of de novo genomic variation. Am J Hum Genet, 2015, 97(1): 67-74.
doi: 10.1016/j.ajhg.2015.05.008 pmid: 26054435 |
| [27] |
Qin L, Wang J, Tian X, Yu H, Truong C, Mitchell JJ, Wierenga KJ, Craigen WJ, Zhang VW, Wong LJC. Detection and quantification of mosaic mutations in disease genes by next-generation sequencing. J Mol Diagn, 2016, 18(3): 446-453.
doi: S1525-1578(16)00039-8 pmid: 26944031 |
| [28] | Gambin T, Liu Q, Karolak JA, Grochowski CM, Xie NG, Wu LR, Yan YH, Cao Y, Coban Akdemir ZH, Wilson TA, Jhangiani SN, Chen E, Eng CM, Muzny D, Posey JE, Yang YP, Zhang DY, Shaw C, Liu PF, Lupski JR, Stankiewicz P. Low-level parental somatic mosaic SNVs in exomes from a large cohort of trios with diverse suspected Mendelian conditions. Genet Med, 2020, 22(11): 1768-1776. |
| [29] | Sim JCH, White SM, Lockhart PJ. ARID1B-mediated disorders: Mutations and possible mechanisms. Intractable Rare Dis Res, 2015, 4(1): 17-23. |
| [30] | van Ravenswaaij-Arts C, Martin DM. New insights and advances in CHARGE syndrome: Diagnosis, etiologies, treatments, and research discoveries. Am J Med Genet C Semin Med Genet, 2017, 175(4): 397-406. |
| [31] | Boothe M, Morris R, Robin N. Stickler syndrome: a review of clinical manifestations and the genetics evaluation. J Pers Med, 2020, 10(3): 105. |
| [32] | Aldè M, Cantarella G, Zanetti D, Pignataro L, La Mantia I, Maiolino L, Ferlito S, Di Mauro P, Cocuzza S, Lechien JR, Iannella G, Simon F, Maniaci A. Autosomal dominant non-syndromic hearing loss (DFNA): a comprehensive narrative review. Biomedicines, 2023, 11(6): 1616. |
| [33] |
Pauli RM. Achondroplasia: a comprehensive clinical review. Orphanet J Rare Dis, 2019, 14(1): 1.
doi: 10.1186/s13023-018-0972-6 pmid: 30606190 |
| [34] | Wang L, Lin QF, Wang HY, Guan J, Lan L, Xie LY, Yu L, Yang J, Zhao C, Liang JL, Zhou HL, Yang HM, Xiong WP, Zhang QJ, Wang DY, Wang QJ. Clinical auditory phenotypes associated with GATA3 gene mutations in familial hypoparathyroidism-deafness-renal dysplasia syndrome. Chin Med J (Engl), 2017, 130(6): 703-709. |
| [35] | Li XH, Huang SS, Wang GJ, Kang DY, Han MY, Wu XD, Yang JY, Zheng QC, Zhao CY, Yuan YY, Dai P. Quantitative assessment of low-level parental mosaicism of SNVs and CNVs in Waardenburg syndrome. Hum Genet, 2023, 142(3): 419-430. |
| [36] | Ruiz-Pérez MV, Henley AB, Arsenian-Henriksson M. The MYCN Protein in health and disease. Genes (Basel), 2017, 8(4): 113. |
| [37] | Gregory GE, Munro KJ, Couper KN, Pathmanaban ON, Brough D. The NLRP3 inflammasome as a target for sensorineural hearing loss. Clin Immunol, 2023, 249: 109287. |
| [38] |
Wang HY, Guan LP, Wu XN, Guan J, Li J, Li N, Wu KL, Gao Y, Bing D, Zhang JG, Lan L, Shi T, Li DY, Wang WJ, Xie LY, Xiong F, Shi W, Zhao LJ, Wang DY, Yin Y, Wang QJ. Clinical and genetic architecture of a large cohort with auditory neuropathy. Hum Genet, 2024, 143(3): 293-309.
doi: 10.1007/s00439-024-02652-7 pmid: 38456936 |
| [39] | Gao X, Huang SS, Qiu SW, Su Y, Wang WQ, Xu HY, Xu JC, Kang DY, Dai P, Yuan YY. Congenital sensorineural hearing loss as the initial presentation of PTPN11- associated Noonan syndrome with multiple lentigines or Noonan syndrome: clinical features and underlying mechanisms. J Med Genet, 2021, 58(7): 465-474. |
| [40] | Wang SQ, Chen Y, Luo KH, Shi NJ, Xiao KL, Cui ZH, Zeng TS, Li HQ. Diagnosis and genetic analysis of a case of Waardenburg syndrome type 2 with hypogonadotropic hypogonadism caused by SOX10 gene deletion. Hereditas(Beijing), 2022, 44(12): 1158-1166. |
| 王思琪, 陈阳, 罗宽宏, 史宁杰, 肖康丽, 崔振海, 曾天舒, 黎慧清. 一例SOX10基因缺失所致的Waardenburg综合征2型合并低促性腺激素性性腺功能减退症的诊断和基因检测分析. 遗传, 2022, 44(12): 1158-1166. |
| [1] | 田盼辉, 许玥, 张永清, 王天云. 遗传病并不一定会遗传:遗传病概念的中文翻译与建议[J]. 遗传, 2024, 46(9): 673-676. |
| [2] | 张秀泉,王建,熊符,吕伟标,周远青,杨少民,张玉婷,田小燕,连蔚,徐湘民. 染色体10q24.31片段重复导致先天性缺指/缺趾畸形的一个家系致病机理分析[J]. 遗传, 2019, 41(8): 716-724. |
| [3] | 孙丽雅, 邢清和, 贺林. 中国出生缺陷遗传学研究的回顾与展望[J]. 遗传, 2018, 40(10): 800-813. |
| [4] | 廖亚平,王春景,梁猛,胡小梅,吴琦. 平衡复杂染色体重排携带者的遗传与生育情况分析[J]. 遗传, 2017, 39(5): 396-412. |
| [5] | 张喆,李巍. 遗传咨询问答(27)[J]. 遗传, 2010, 32(9): 0-0. |
| [6] | 张喆,李巍. 遗传咨询问答(26)[J]. 遗传, 2010, 32(3): 0-0. |
| [7] | 贺敏,李巍. 中国遗传咨询网——我国首个在线遗传咨询与遗传教育网站的开发[J]. 遗传, 2007, 29(3): 381-381―384. |
| [8] | 章远志,Nanbert Zhong. 心理治疗模式遗传咨询的工作重点及基本程序[J]. 遗传, 2006, 28(11): 1440-1444. |
| [9] | 王树玉,王素桂,任国庆,贾婵维,马延敏,薛虹. 优生与遗传咨询的临床研究[J]. 遗传, 2002, 24(6): 631-635. |
| [10] | 符生苗,符惠群,钟文玲,程在玉. 335例遗传咨询病理的染色体分析[J]. 遗传, 1990, 12(5): 33-35. |
| [11] | 张晓珍,余继英,霍晓春,饶兆英,柳丽瑰. 826例遗传咨询病人的染色体分析[J]. 遗传, 1990, 12(1): 24-26. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||

www.chinagene.cn
备案号:京ICP备09063187号-4
总访问:,今日访问:,当前在线:
