



近20年来基因组学经历了爆发式的发展,如今已经成为生命科学领域研究的重要范畴。基因组承载着生命体的基本遗传信息,一个高质量的基因组是展开深度遗传学及分子功能研究的先决条件。然而,随着基因组学理论体系的延展、测序技术的革新、数据维度和数据需求的不断丰富,研究者对基因组本身的认知经历了不断的扩充与迭代。将单一的参考基因组作为特定物种或者类群基因组的“标准品”,其代表性和蕴含的生物多样性始终是有限的。物种内、种系间的差异是解析种群演化和表型特征形成的关键,不能被忽视。针对这些问题,研究人员不断探索新的研究方法与思路,这此过程中考虑多个代表性基因组比较与整合的泛基因组学(pan-genomics)框架得以建立,成为现今研究的热点方向。
作物分子设计育种是解决国家粮食安全问题的重要手段,而高质量的作物基因组是遗传学家、育种家认识改造作物的关键基础。作物基因组演化存在诸多特征。一方面,植物基因组中基因组序列重复、基因组加倍、多倍化等事件更为频繁,使得植物在染色体水平上积累了更多的结构差异[1];另一方面,作物驯化改良是一致性和多样化兼有的过程,尽管品种/品系之间具备高度的可比性,但单个品种/品系的基因组并不能代表整个作物的遗传背景。因此研究者认识到,使用单个基因组作为参考开展作物遗传与功能基因组研究,很可能低估研究对象遗传分化的程度并遗失诸多重要的遗传变异[2,3]。以上特征表明作物是开展泛基因组研究的良好素材,而泛基因组也是深度解析作物基因组多样性、挖掘农艺性状相关位点的重要方法。作为传统基因组形式的补充和扩展,泛基因组现今已成为作物基因组图谱绘制和遗传解析的常用手段[4,5]。
大豆(Glycine max)是我国重要的作物和经济物资,由于需求的激增导致供给不足,国内大豆不得不大量依赖进口。改良种质,培育高产、稳产、高品质、适应不同农田环境的大豆,是提高大豆产量的关键。中国拥有最丰富的大豆遗传资源以及多样的栽植生态区系,采用泛基因组的研究方法,厘清大豆的遗传变异,发掘新的或未被充分使用的遗传位点,结合分子设计育种等手段,对于推进中国大豆品种的选优改良,具有重要意义。
1 泛基因组概述
1.1 泛基因组概念的发展
泛基因组(pan-genome)的词缀“pan”来源于希腊语,意为“全”、“一切”。泛基因组通常意义上是指代一个物种/类群所有基因组,或代表性基因组的总和。在研究的早期,测序技术产出的数据质量有限,测序成本高昂,在许多真核生物中获得单个高质量组装基因组是十分困难的事情。因此,往往用单个或少数高完成度的基因组作为一个物种或是一个类群的代表或参考。而在一些原核生物中,由于基因组规模小,获取基因组相对容易,研究人员通常可以获得同一个类群中多个个体的完整基因组,并且开展多基因组间的整体比对。这类工作最早由Tettelin等[6]于2005年在无乳链球菌(Streptococcus agalactiae)中开展,是泛基因组研究的雏形。
然而泛基因组的概念推广到更复杂的动植物等真核生物类群并没有那么迅速。首先,通常情况下真核生物基因组相比细菌要大得多,这意味着基因组测序的成本和后续组装消耗的算力、时间资源都很巨大。其次,真核生物基因组更为复杂,多倍体、高重复序列、高杂合度等情况都会增加基因组组装的难度[7⇓⇓~10]。并且由于基因组成分复杂,有大量非基因区序列、重复序列的存在,使得泛基因组组分评估及基因组差异的鉴定也不易进行[11]。近几年,随着测序技术的发展,测序成本下降,比较基因组学手段不断完善,这些问题才逐渐得到解决。从原核生物到真核生物,泛基因组的范畴也从包含全体注释基因扩展到包含所有基因组序列。而伴随组学研究维度的开拓,泛组学概念的应用也从基因组层面延伸到如泛转录组、泛三维基因组等层面[12,13]。
1.2 泛基因组研究的核心问题
泛基因组研究的核心问题,是对物种/类群基因组完备性或者代表性遗传信息的描述[14]。与群体遗传学类似,泛基因组的研究对象并非单一个体。然而群体遗传学层面的基因组研究侧重于发掘变异位点及遗传多态性,即个体间的异质性。而个体间的异质性和同质性,即共享与差异的基因组成分,均为泛基因组研究描述的内容。通过泛基因组研究,人们能了解一个物种/类群的完整基因组架构,并借此推断构成这一物种/类群的核心遗传信息(即基因组下界),以及物种/类群的遗传分化程度(即基因组上界)。
此外,泛基因组研究涉及基因组间的比较和整合,其中对不同基因组间染色体结构变异(structural variation,SV)的挖掘和处理也成为研究的重要环节[15]。相较于单核苷酸多态性(single nucleotide polymorphism,SNP),结构变异的长度不定,变异类型更为复杂,处理难度也更高。同时,结构变异引起的基因组改变更为剧烈,更易引起物种间表型特征的多态性。这类变异在基因组学研究的早期,因为技术和成本的限制,很难作为重要的研究方向,而如今则成为泛基因组研究聚焦的重点之一。对于染色体结构变异的处理,也体现了泛基因组实践策略的不同发展阶段。
2 泛基因组实践策略及研究实例
2.1 从头组装/比对组装基因组
这类方法在实践层面上最为简单,在泛基因组研究的早期有较多应用,但也存在诸多问题。单独基因组形式的泛基因组通常包含过多冗余的数据量和数据维度。而“参考基因组+额外序列”的方式对于泛基因组的组织并不直观有效。因此研究者需要探索更为高效合理的泛基因组数据组织形式。
2.2 迭代式泛基因组
迭代式泛基因组相较于从头组装的泛基因组整合度高,不引入额外序列,并且类似传统的线性基因组,更易于理解。但实现过程中对于原有基因组的覆盖将不可避免丢失许多单独基因组状态下的特征。因此,迭代式组装尽管减少了信息的冗余,也同时存在大量的信息丢失[11]。
2.3 基于图论的泛基因组
尽管图基因组并不像传统线性基因组那样直观,但其最大程度压缩了冗余信息,并且保留了有义信息。此外图基因组可以灵活地进行数据组合与还原,保证了组学数据的可读性。对于基因组较大,变异复杂的真核生物,图基因组是更适合的方法,也成为现在的趋势[24⇓⇓⇓~28]。此外,图基因组更兼容计算机的I/O形式,能够更快、更有效地进行基于二代测序数据的比对和结构变异检测。目前,图基因组是泛基因组数据存储、调用、展示等综合性能最佳的形式,越来越多的基因组分析工具开始向该方向发展,如vg (Variation Graph toolkit)[26]、GraphTyper2[25]、Giraffe[29]、odgi (Optimized Dynamic Genome/Graph Implementation)[30]、pggb (PanGenome Graph Builder)[31]等。一些经典的工具,如HISAT2[32]也有此方面功能的拓展。图基因组在泛基因组,尤其是植物泛基因组学领域,目前已经有了很多实践,逐渐成为研究的主流方法。
2.4 作物泛基因组研究
2011年,Gan等[33]对拟南芥(Arabidopsis thaliana)自然群体材料的基因组比较是植物泛基因组研究的开端。该工作从头组装了18个拟南芥的单拷贝序列基因组,通过比较发现了相对参考基因组共有28.3 Mb非冗余变异序列,平均每个样品4.5~7.6 Mb。此后泛基因组研究逐渐在植物中兴起,并且在近10年间高速发展。目前许多植物,特别是作物都完成了从单一参考基因组到泛基因组的整合与跨越[20,22,34⇓⇓⇓⇓~39]。早期植物泛基因组多采用从头组装/比对组装的策略进行构建,部分研究采用了迭代组装方式(表1)。在近期的研究中,从头组装结合图泛基因组已经成为主流的泛基因组研究策略(表1)。泛基因组研究在一定程度上揭示了作物物种内或近缘种间的基因组变异规模。对比一些研究结果可以得出,在不同植物类群的泛基因组中,核心基因家族占总基因家族数量的40%~70%,表明30%~60%的基因家族在物种内发生了获得/丢失的变异[16,17,19⇓⇓~22,40,41]。
表1 植物泛基因组研究实例汇总
Table 1
| 类群 | 发表年份 | 样品数 | 测序方式 | 泛基因组构建策略 | 参考文献 |
|---|---|---|---|---|---|
| 拟南芥(Arabidopsis thaliana) | 2011 | 18 | 二代测序 | 迭代组装+从头组装 | [33] |
| 野生大豆(Glycine soja) | 2014 | 7 | 二代测序 | 从头组装 | [17] |
| 甘蓝(Brassica oleracea) | 2016 | 9 | 二代测序 | 迭代组装 | [22] |
| 苜蓿(Medicago truncatula) | 2017 | 15 | 二代测序 | 从头组装 | [76] |
| 二穗短柄草(Brachypodium distachyon) | 2017 | 54 | 二代测序 | 从头组装 | [16] |
| 水稻(Oryza sativa) | 2018 | 3010 | 二代测序+三代测序 | 比对组装 | [21] |
| 野生及栽培水稻(O. rufipogon, O. sativa) | 2018 | 66 | 二代测序 | 比对组装 | [42] |
| 水稻属及亲缘物种(Oryza, Leersia) | 2018 | 13 | 三代测序+二代测序 | 从头组装 | [18] |
| 辣椒属(Capsicum) | 2018 | 168 | 二代测序 | 比对组装 | [77] |
| 芝麻(Sesamum indicum) | 2018 | 5 | 二代测序 | 比对组装 | [78] |
| 番茄及野生亲缘种(Solanum section Lycopersicon) | 2019 | 725 | 二代测序 | 比对组装 | [19] |
| 向日葵(Helianthus annuus) | 2019 | 287 | 二代测序 | 比对组装 | [20] |
| 油菜(Brassica napus) | 2020 | 8 | 三代测序 | 从头组装 | [43] |
| 野生及栽培大豆(Glycine subgenus Soja) | 2020 | 29 | 三代测序 | 从头组装+图基因组 | [39] |
| 大麦(Hordeum vulgare) | 2020 | 20 | 二代测序+三代测序 | 从头组装 | [79] |
| 番茄及野生亲缘种(Solanum section Lycopersicon) | 2020 | 14 | 二代测序+三代测序 | 比对组装(泛结构变异) | [45] |
| 鹰嘴豆(Cicer arietinum) | 2021 | 3366 | 二代测序 | 比对组装 | [80] |
| 棉花及亲缘种(Gossypium) | 2021 | 1961 | 二代测序 | 比对组装 | [81] |
| 野生及栽培高粱(Sorghum bicolor) | 2021 | 13 | 三代测序 | 从头组装 | [82] |
| 玉米(Zea may) | 2021 | 26 | 三代测序 | 从头组装 | [83] |
| 水稻(O. sativa) | 2021 | 33 | 三代测序 | 从头组装+图基因组 | [34] |
| 野生及栽培萝卜(Raphanus) | 2021 | 11 | 三代测序 | 从头组装+图基因组 | [84] |
| 黄瓜 (Cucumis sativus) | 2022 | 12 | 三代测序 | 从头组装+图基因组 | [38] |
| 水稻属(Oryza) | 2022 | 251 | 三代测序 | 从头组装+图基因组 | [85] |
| 棉花属(Gossypium) | 2022 | 10 | 三代测序 | 从头组装+图基因组 | [86] |
| 多年生大豆(Glycine subgenus Glycine) | 2022 | 6 | 三代测序 | 从头组装 | [62] |
| 野生及栽培马铃薯(Solanum section Petota) | 2022 | 44 | 三代测序 | 从头组装 | [87] |
| 番茄(Solanum lycopersicum) | 2022 | 32 | 三代测序 | 从头组装+图基因组 | [35] |
| 野生及栽培谷子(Setaria) | 2023 | 110 | 三代测序 | 从头组装+图基因组 | [40] |
| 茶(Camellia sinensis) | 2023 | 22 | 三代测序 | 从头组装+图基因组 | [41] |
| 柑橘属(Citrus) | 2023 | 12 | 三代测序 | 从头组装+图基因组 | [36] |
| 番茄及野生亲缘种(Solanum section Lycopersicon) | 2023 | 13 | 三代测序 | 从头组装+图基因组 | [85] |
| 玉米(Z. mays) | 2023 | 12 | 三代测序 | 从头组装 | [88] |
| 野生及栽培黍(Panicum miliaceum) | 2023 | 32 | 三代测序 | 从头组装+图基因组 | [46] |
泛基因组是深度挖掘农艺性状与基因组变异,尤其是染色体结构变异关联性的有效手段。一方面,对于已知基因或位点,泛基因组能够提供更新、更全面的变异认知。野生大豆(Glycine soja)的泛基因组研究比较了大豆开花途径基因的变异,发现PHY4、E3、E4、E1、FT、LFY等基因在野生及栽培大豆基因组间均存在蛋白差异,并且FT在野生大豆中存在一个参考基因组WM82中没有的亚型[17]。这些变异可能导致了野生和栽培大豆开花特征的分化。66份野生和栽培水稻的泛基因组研究充分挖掘了waxy、Hd1等位点的多种单倍型,涉及SNP和Indel的多种组合,加深了对水稻品质、花期等复杂农艺性状的理解[42]。谷子(Setaria italica)泛基因组研究表明,种质间落粒性、籽粒大小差异与染色体结构变异相关。其中,在其他谷物中被平行选择的sh1基因,在谷子中也发生了一个855 bp的存在/缺失变异(presence and absence variation,PAV),造成基因的获得/缺失,进而控制落粒性的变化[40]。这也体现出sh1在谷物中功能的保守性和利用改造价值。另一方面,群体结构变异数据可以用作关联分析,发挥和SNP相当或者互补的效力。Song等[43]在油菜(Brassica napus)泛基因组研究中使用PAV数据进行种子重量的全基因组关联分析(genome wide associated study,GWAS),其信号区间和使用SNP的计算结果重叠,而其中一个3.6 kb的PAV位于信号峰值。该变异为转座元件(transposable element,TE)插入,统计NAM群体的表型发现该变异的存在/缺失和角果长度和种子重量都显著相关。而该TE下游为CYP78A9基因,推测变异影响了该基因的表达,从而造成性状的变化。谷子泛基因组研究中对千粒重、粒宽的SV-GWAS分析找到一个控制相关表型的基因及变异位点[40]。该基因启动子区发生了366 bp的PAV。实验表明,该序列变异导致基因表达量改变,相关过表达株系也表现出粒宽的显著下降。水稻中对于产量的分析发现,使用结构变异进行GWAS分析能够检测到比SNP更为显著的关联位点,其中位于OsNPY2基因上游的一个1.4 kb序列存在/缺失与产量表型密切关联[44]。
3 大豆泛基因组研究
3.1 大豆属泛基因组组成
2014年野生大豆的泛基因组研究是植物中第一项明确泛基因组概念的工作[17]。然而其数据质量、全面性和挖掘深度都受到了时代和技术的制约。2020年一项包含大豆属Soja亚属的野生、栽培大豆在内,26个大豆种质材料基因组、转录组及近3000份种质材料重测序的工作则更精准地描绘了大豆的遗传变异图谱,系统阐述了染色体结构变异在大豆演化/驯化中发挥的作用[39]。该研究从2898份来自世界大豆主要栽植区的种质资源中共检测到约3千万个单核苷酸变异位点。根据系统发育关系,挑选出26个代表性的种质,进行基因组从头组装和泛基因组构建。这26个种质按类群划分包括野生、农家种、栽培品种,按用途划分包括骨干亲本及区域主栽品种等,从头组装基因组大小在992.3~1059.8 Mb之间,样品序列锚定在染色体上的比率平均为99.0%,二代测序比对回自身基因组的比对率平均在99.4%。基因组重复序列注释检测到大豆基因组的平均重复序列比例为54.4%,蛋白编码基因注释表明大豆泛基因组样品平均注释基因数量为56,522,BUSCO检验平均达到95.6%。以上结果符合大豆基因组的基本特征,说明基因组组装注释质量达到高水平。
对26个大豆从头组装基因组,连同已经报道的ZH13的基因组进行基因家族聚类,所有基因被分入57,492个基因家族,这与之前野生大豆中报道的数量接近[17]。对不同品种数量构建的泛基因、核心基因家族数目的抽样统计显示,泛基因组的数量在25个样品时到达了平台期,意味着该研究的取样对于大豆基因组已具有足够的代表性。将基因家族按样品出现的频数作为划分,得到大豆的核心基因家族(频数为27) 20,623个,松弛核心基因家族(频数为25、26) 8163个,非必需基因家族(频数为2~24) 28,679个,私有基因家族(频数为1) 27个。由此得出,大豆泛基因组中核心(及松弛核心)基因家族占总基因家族的50.1%,非必需及私有家族(可变家族)的数量占49.9%。该结果符合以往研究得出的植物中30%~ 60%的基因家族为可变家族的认知[16,17,19⇓⇓~22,40,41]。
3.2 大豆属泛基因组变异
泛基因组包含的变异是否能反应物种群体水平的变异,是值得探讨的问题。以ZH13基因组作为参考,结合26个泛基因组样品和已报道的WM82及W05的基因组数据,在29个大豆基因组上检测到14,604,953个SNP和12,716,823个Indel (≤50 bp)[39]。该数据与2898份重测序的变异数据进行比较,尽管SNP数量比2898份重测序要少,但是二者分布特征相似。以500 kb区间为窗口进行全基因组扫描,过滤2898份重测序中次等位基因频率(minor allele frequency,MAF)<0.01的位点后,其与29个基因组中SNP数量的皮尔森相关性系数为0.553。此外π、dN/dS等群体遗传学特征值在29个基因组与2898份重测序数据间同样具有很高的相关性。这表明泛基因组对于变异的检测具有群体水平的代表性。
大尺度结构变异(>50 bp)采用短序列测序方式往往很难鉴定。通过基因组比对的方式,以ZH13为参考在28个大豆基因组中检测到共计776,399个结构变异,其中723,862个PAV、27,531个拷贝数变异(copy number variation,CNV)、21,886个易位事件、3120个倒位事件[39]。PAV的长度主要分布在1~2 kb,易位长度主要分布在10~30 kb,倒位长度主要分布在100~200 kb。CNV的变化倍数主要在2~3倍。泛基因组中检测到的723,862个PAV共计4.71 Gb序列长度,平均每个样品167.09 Mb,占基因组大小约16%。比较每个样品的获得与缺失序列长度之差,及其与ZH13基因组大小之差,发现二者具有很高的相关性,说明PAV是造成样品间基因组大小差异的主要来源。在大豆中结构变异在基因组重复序列区域显著富集,其中78.5%的PAV来自于DNA重复。对番茄(Solanum lycopersicum)泛基因组研究发现84%的序列缺失与76%的序列插入变异与重复序列重合(>100 bp)[45]。对黍(Panicum miliaceum)的泛基因组研究发现PAV与TE的重合比例在70%左右[46]。这些结果暗示一些植物中序列重复事件可能是结构变异发生的重要驱动力,进而导致物种内基因组大小的波动。
3.3 大豆属图泛基因组构建
图1
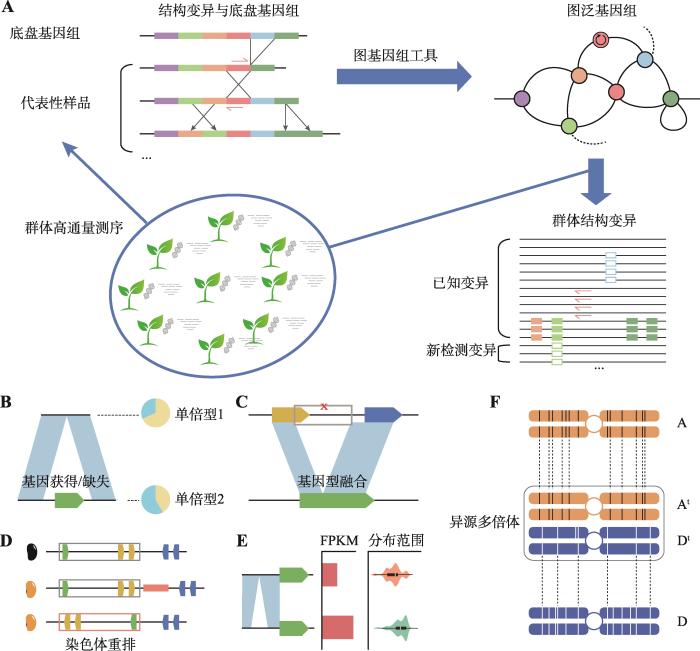
图1
作物泛基因组研究策略及认知
A:图泛基因组研究基本流程,包括群体测序筛选代表性样品、结构变异分析、图泛基因组构建、群体结构变异检测等;B~E:泛基因组视角下的大豆农艺性状、演化历程遗传机制认知,包括基因获得/缺失与种皮亮度(B)、基因融合与E3基因多态性(C)、染色体重排与种皮颜色(D)、结构变异对基因表达调控与种质分布(E);F:异源多倍体大豆的冗余基因丢失与亚基因组偏好性。
Fig. 1
Crop pan-genome strategy and knowledge
此外,研究表明将结构变异中重复序列占总长度90%的条目过滤,是有效的数据压缩、降低错误率的策略。Liu等[39]采用vg工具,以过滤后的结构变异数据为输入,ZH13基因组为底盘基因组,构建可用于检索和二代数据比对的大豆图泛基因组索引文件。将2898个大豆样品重测序数据比对到图泛基因组上,共计检测到55,402个结构变异。采用图泛基因组检测结构变异的精确率、召回率及F-score分别为0.94、0.75和0.83,表明图泛基因组结合群体二代测序数据是作物中进行大规模结构变异检测的可行方法。图泛基因组流程检测的结构变异N50为659/595 bp(缺失/插入),远高于GATK流程的3/3 bp,说明图泛基因组流程对于大尺度结构变异检测具有很好的效力。相对于28个基因组中检测到的变异,在约3000份群体水平找到3584个新的结构变异,占总变异数的6.5%,并且这些变异的出现频率较低。野生大豆中检测到的已有和新结构变异的数量均明显高于农家种和栽培大豆。水稻中相似研究检测到的新结构变异占总变异数的16.4%[34],但该研究的图泛基因组构建仅针对栽培稻进行。这也侧面反应出作物的野生种可能持有更丰富的变异类型,在作物泛基因组研究中加入野生类群可以很好地提升遗传变异的覆盖度。
3.4 泛基因组助力大豆演化/驯化遗传基础
位于基因区的结构变异可能造成基因开放阅读框(open reading frame,ORF)的改变,进而导致功能的丢失或分化。其中结构变异造成的转录本通读是一种较为特殊的情况,即由于序列丢失导致原本独立转录的基因融合为一个转录本。转录本通读引起的基因融合在基因进化过程中起到重要作用[48]。依赖大规模的泛基因组数据,不仅能确认已有报道的等位基因,也能鉴定包括融合基因在内的基因新结构(图1C),例如大豆开花相关的主效基因E3[49]。自然状态下,E3以复等位基因的形式存在[50]。26个从头组装基因组的注释基因与ZH13的E3进行比较,可以找到一个从E3第3个内含子开始的13.3 kb缺失。该变异造成了其中一个基因(SoyZH13_ 19G210500)的完全丢失[39]。RNAseq数据显示该变异除了导致E3的最后一个外显子及SoyZH13_ 19G210500的缺失外,还造成了E3和SoyZH13_ 19G210600的转录本读通。此外,该变异还造成了E3在缺失最后一个外显子后获得了一个额外的外显子。PCR片段测序验证了E3与SoyZH13_19G210600的基因融合事件,以及外显子改变事件是真实存在且相互独立的。泛基因组挖掘并验证了E3基因由结构变异产生的大量多态性,包括基因融合与ORF改变等,这可能是塑造大豆区域适应性分化的重要原因。
大豆的许多性状控制遗传位点,由于变异类型复杂、涉及基因多而难以被克隆[17,51⇓⇓~54]。大规模从头组装的基因组使得这类解析变得可能(图1D)。大豆种皮颜色相关的I位点是受驯化位点[54,55],使大豆种皮从黑色转变为黄色。该位点为一系列异黄酮代谢途径中查尔斯酮合成酶(CHS)基因组成的基因簇,存在同源依赖的基因沉默(homology dependent gene silencing,HDGS)机制,调控CHS基因的表达[56⇓~58]。Liu等[39]在29个大豆基因组中调查种皮颜色的表型以及I位点,发现4个野生大豆和农家种SoyL02表现为黑色种皮,其余栽培大豆均为黄色种皮。I位点及周边的SNP构建系统发育树发现黑或黄种皮的样品各自聚类在一起。结构变异分析表明,相对于黑种皮类型基因组,一部分黄种皮样品的基因组上存在一个约100 kb的倒位以及CHS序列单元的重复,这与之前的报道相符[59]。然而另一部分样品中,虽然这个约100 kb的倒位变异不存在,仍然表现出黄色种皮。尽管如此,其上有一段约23 kb的序列发生了重复,并且插入到其后的CHS反向重复基因簇中,而这很可能导致了双交换事件并造成周围CHS单元的假基因化。因此,I位点周围的染色体变异得到完整的解析,而调控机制有待于进一步探索。
基因表达可能受到基因附近调控区序列变异的影响,进而导致农艺性状的变化。泛基因组结合转录组的研究策略能够深入挖掘由染色体结构变异导致的表达量差异,从而定位农艺性状的候选基因和变异(图1E)。缺铁萎黄是大豆在石灰土中种植时常见的病症。Lin等[60]的研究已定位到若干与铁离子利用效率相关的QTL位点,其中一个位于14号染色体。该QTL中存在一个注释为铁/锌离子调控转运蛋白的基因SoyZH13_14G179600,其5′启动子区在泛基因组中检测到一个1.4 kb的PAV[39]。该PAV满足转座子DNA Mutator的序列特征[61],并且可以将26个大豆种质分成两组:未发生序列缺失和发生序列缺失的类型。RNA-seq数据表明,后者相对前者具有更高的表达量。结合群体基因型数据和样品信息记录发现,1.4 kb序列缺失的样品主要分布在纬度更高的种植区,而未发生序列缺失样品分布在纬度较低的地理区域。中国不同地理区域的土壤pH不同,进而影响铁离子浓度。因此,区域差异可能是造成遗传分化的诱因。
3.5 多年生大豆泛基因组研究
大豆属除了分布于东亚地区的一年生大豆(Soja亚属)之外,还有约30个分布于澳大利亚的多年生大豆物种(Glycine亚属)。该类群虽然和栽培大豆分化较大,但是部分物种染色体数目与栽培大豆相同,可能是栽培大豆潜在的遗传改良基因资源库,具有研究价值。2022年,一项针对Glycine亚属6个物种(5个二倍体和1个四倍体)的泛基因组研究系统地揭示了多年生大豆的基因组演化特征[62]。二倍体物种基因组大小为935.6~1373.8 Mb,平均大小1105 Mb左右,与Soja亚属大致接近,而基因组预测的蛋白质编码基因有70%在一年生大豆中缺失。多年生大豆相对栽培大豆而言,整体基因组变异幅度较大,遗传资源应用可能更侧重于定向基因改造或替换而非远源杂交。
以菜豆(Phaseolus vulgaris)为参考的比较基因组发现,多年生大豆相对于一年生大豆,基因组重排事件更少,染色体更为稳定。Zhuang等[62]研究计算了同源基因家族在一年生、多年生大豆中的Ka/Ks,发现52个家族在两个亚属中发生了净化选择;其中PHP、D14等是与开花、植株发育相关的基因,在两个亚属内计算Ka/Ks值低,但是在亚属间计算则有较高的Ka/Ks值,暗示这些基因可能参与了亚属间生活史策略的分化。
4 结语与展望
4.1 未来泛基因组发展
测序技术在过去的40年间飞速发展,积累了海量的数据,包括大规模群体测序和从头组装基因组。在此基础上,泛基因组学应运而生,并且受到学界越来越多的重视[4,14,65⇓⇓⇓⇓~70],成为作物遗传育种研究的“利器”[35,40,71]。水稻、玉米、大豆、番茄等作物中不断有泛基因组研究涌现,这些结果或展示了不同研究类群框架下的基因组差异特征,或随着研究技术的提升给出了更高质量的组学参考数据。泛基因组作为一种基于比较基因组的研究方式,研究对象的选择尤为关键。应根据研究目的划定适合的类群范围,挑选代表性个体。泛基因组构建策略的选择应根据样品数量、测序成本以及最终期望呈现的数据结果综合考虑。图泛基因组作为当下泛基因组研究的前沿和热点,整合构建图泛基因组的算法和软件逐渐多样成熟,但这些算法软件多针对人类泛基因组的研究开发。目前植物研究中主要的泛基因组构建策略多是通过三代测序获得高质量的从头组装染色体水平基因组,再借由比较基因组分析结构变异构建图泛基因组。而图泛基因组本身并不依赖除底盘基因组外其他样品的染色体水平基因组组装,因此,三代测序直接检测结构变异结合底盘基因组构建图泛基因组的方法可能是更低成本及更便利的一种方式。此外,针对植物基因组特征,开发解决重复序列比例大、染色体结构变异复杂、基因组大小差异显著的算法和软件,将能够有效提升植物图泛基因组的精度和构建效率。
4.2 多维组学数据应用
大数据时代下,新的数据类型不断涌现,其应用和处理场景也日趋复杂。泛基因组研究通常会在一个物种/类群内产生多套参考基因组数据。建立这些基因组间的关联,高效地进行多基因组的联合检索和调用,是后基因组时代迫切的数据需求。图泛基因组是对这类问题很好的回答,但也带来了新的挑战。首先图基因组是与以往不同的数据形式,针对这类数据开发的数据库和前端应用目前仍然有限。如何将这些数据高效地服务于更多研究者,是值得探索的方向。大豆多维组学数据库SoyOmics对图泛基因组的单倍型检索和数据可视化提供了实践参考[74]。此外,全景多维组学的发展,对于当下数据的提炼和整合能力有了更深的要求。通过多维组学数据的联合应用,提升生物信息学分析结果的精度和可信度,从而提高作物遗传解析效率,最终服务于分子设计育种[75]。在此过程中,针对多层次组学信号的联合处理与评估,以及多层次组学数据网络的构建,应该成为未来探索的重要方向。
在后基因组时代,泛基因组能够起到对传统基因组的补充和发展作用,其价值和必要性已被证实。在大豆中,泛基因组、变异组、转录组、表观组、表型组等多维度数据已有充分的积累。未来的遗传育种研究应当利用好这些多维组学数据,深度解析重要农艺性状的遗传网络,为分子设计育种提供有力指导,这也是提升大豆产量、改善大豆品质的重要路径。
(责任编委: 孔凡江)
中国科学院遗传与发育生物学研究所田志喜课题组简介
中国科学院遗传与发育生物学研究所田志喜课题组成立于2010年。研究团队致力于大豆功能基因组研究和品种培育,以“中华大豆之崛起”为己任,在多维组学立体解析、农艺性状分子机制挖掘、分子育种等方面开展了全方位系统性的工作,取得了一系列重要理论和实践成果。在Cell、Nature Biotechnology、Nature Genetics、Nature Communication、PNAS、Genome Biology、Molecular Plant、Plant Cell、Plant Biotechnology Journal等期刊上发表论文90篇,总引用12,000余次,h指数39。其中ESI高被引论文14,平均单篇他引123.39次。多次应邀在Current Opinion in Plant Biology、Molecular Plant等期刊上撰写综述、评论文章。申请专利7项,培育新品种9个。团队承担科技部、农业部、国家自然科学基金委及中国科学院的一系列重大项目。获评2023年第四届中国科学院“科苑名匠”。

参考文献
Whole-genome duplication and plant macroevolution
DOI:S1360-1385(18)30159-6
PMID:30122372
[本文引用: 1]

Whole-genome duplication (WGD) is characteristic of almost all fundamental lineages of land plants. Unfortunately, the timings of WGD events are loosely constrained and hypotheses of evolutionary consequence are poorly formulated, making them difficult to test. Using examples from across the plant kingdom, we show that estimates of timing can be improved through the application of molecular clock methodology to multigene datasets. Further, we show that phenotypic change can be quantified in morphospaces and that relative phenotypic disparity can be compared in the light of WGD. Together, these approaches facilitate tests of hypotheses on the role of WGD in plant evolution, underscoring the potential of plants as a model system for investigating the role WGD in macroevolution.Copyright © 2018 Elsevier Ltd. All rights reserved.
Plant pangenomics: approaches, applications and advancements
DOI:S1369-5266(19)30120-7
PMID:31982844
[本文引用: 3]
With the assembly of increasing numbers of plant genomes, it is becoming accepted that a single reference assembly does not reflect the gene diversity of a species. The production of pangenomes, which reflect the structural variation and polymorphisms in genomes, enables in depth comparisons of variation within species or higher taxonomic groups. In this review, we discuss the current and emerging approaches for pangenome assembly, analysis and visualisation. In addition, we consider the potential of pangenomes for applied crop improvement, evolutionary and biodiversity studies. To fully exploit the value of pangenomes it is important to integrate broad information such as phenotypic, environmental, and expression data to gain insights into the role of variable regions within genomes.Copyright © 2019. Published by Elsevier Ltd.
Structural variations in plant genomes
DOI:10.1093/bfgp/elu016
PMID:24907366
[本文引用: 1]
Differences between plant genomes range from single nucleotide polymorphisms to large-scale duplications, deletions and rearrangements. The large polymorphisms are termed structural variants (SVs). SVs have received significant attention in human genetics and were found to be responsible for various chronic diseases. However, little effort has been directed towards understanding the role of SVs in plants. Many recent advances in plant genetics have resulted from improvements in high-resolution technologies for measuring SVs, including microarray-based techniques, and more recently, high-throughput DNA sequencing. In this review we describe recent reports of SV in plants and describe the genomic technologies currently used to measure these SVs. © The Author 2014. Published by Oxford University Press.
Towards plant pangenomics
DOI:10.1111/pbi.12499
PMID:26593040
[本文引用: 3]
As an increasing number of genome sequences become available for a wide range of species, there is a growing understanding that the genome of a single individual is insufficient to represent the gene diversity within a whole species. Many studies examine the sequence diversity within genes, and this allelic variation is an important source of phenotypic variation which can be selected for by man or nature. However, the significant gene presence/absence variation that has been observed within species and the impact of this variation on traits is only now being studied in detail. The sum of the genes for a species is termed the pangenome, and the determination and characterization of the pangenome is a requirement to understand variation within a species. In this review, we explore the current progress in pangenomics as well as methods and approaches for the characterization of pangenomes for a wide range of plant species.© 2015 Society for Experimental Biology, Association of Applied Biologists and John Wiley & Sons Ltd.
Exploring and exploiting pan-genomics for crop improvement
DOI:S1674-2052(18)30383-6
PMID:30594655
[本文引用: 1]
Genetic variation ranging from single-nucleotide polymorphisms to large structural variants (SVs) can cause variation of gene content among individuals within the same species. There is an increasing appreciation that a single reference genome is insufficient to capture the full landscape of genetic diversity of a species. Pan-genome analysis offers a platform to evaluate the genetic diversity of a species via investigation of its entire genome repertoire. Although a recent wave of pan-genomic studies has shed new light on crop diversity and improvement using advanced sequencing technology, the potential applications of crop pan-genomics in crop improvement are yet to be fully exploited. In this review, we highlight the progress achieved in understanding crop pan-genomics, discuss biological activities that cause SVs, review important agronomical traits affected by SVs, and present our perspective on the application of pan-genomics in crop improvement.Copyright © 2019 The Author. Published by Elsevier Inc. All rights reserved.
Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”
DOI:10.1073/pnas.0506758102
PMID:16172379
[本文引用: 1]
The development of efficient and inexpensive genome sequencing methods has revolutionized the study of human bacterial pathogens and improved vaccine design. Unfortunately, the sequence of a single genome does not reflect how genetic variability drives pathogenesis within a bacterial species and also limits genome-wide screens for vaccine candidates or for antimicrobial targets. We have generated the genomic sequence of six strains representing the five major disease-causing serotypes of Streptococcus agalactiae, the main cause of neonatal infection in humans. Analysis of these genomes and those available in databases showed that the S. agalactiae species can be described by a pan-genome consisting of a core genome shared by all isolates, accounting for approximately 80% of any single genome, plus a dispensable genome consisting of partially shared and strain-specific genes. Mathematical extrapolation of the data suggests that the gene reservoir available for inclusion in the S. agalactiae pan-genome is vast and that unique genes will continue to be identified even after sequencing hundreds of genomes.
De novo genome assembly: what every biologist should know
Origin and evolution of the octoploid strawberry genome
DOI:10.1038/s41588-019-0356-4
PMID:30804557
[本文引用: 1]
Cultivated strawberry emerged from the hybridization of two wild octoploid species, both descendants from the merger of four diploid progenitor species into a single nucleus more than 1 million years ago. Here we report a near-complete chromosome-scale assembly for cultivated octoploid strawberry (Fragaria × ananassa) and uncovered the origin and evolutionary processes that shaped this complex allopolyploid. We identified the extant relatives of each diploid progenitor species and provide support for the North American origin of octoploid strawberry. We examined the dynamics among the four subgenomes in octoploid strawberry and uncovered the presence of a single dominant subgenome with significantly greater gene content, gene expression abundance, and biased exchanges between homoeologous chromosomes, as compared with the other subgenomes. Pathway analysis showed that certain metabolomic and disease-resistance traits are largely controlled by the dominant subgenome. These findings and the reference genome should serve as a powerful platform for future evolutionary studies and enable molecular breeding in strawberry.
HaploMerger2: rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly
DOI:10.1093/bioinformatics/btx220
PMID:28407147
[本文引用: 1]
De novo assembly is a difficult issue for heterozygous diploid genomes. The advent of high-throughput short-read and long-read sequencing technologies provides both new challenges and potential solutions to the issue. Here, we present HaploMerger2 (HM2), an automated pipeline for rebuilding both haploid sub-assemblies from the polymorphic diploid genome assembly. It is designed to work on pre-existing diploid assemblies, which are typically created by using de novo assemblers. HM2 can process any diploid assemblies, but it is especially suitable for diploid assemblies with high heterozygosity (≥3%), which can be difficult for other tools. This pipeline also implements flexible and sensitive assembly error detection, a hierarchical scaffolding procedure and a reliable gap-closing method for haploid sub-assemblies. Using HM2, we demonstrate that two haploid sub-assemblies reconstructed from a real, highly-polymorphic diploid assembly show greatly improved continuity.Source code, executables and the testing dataset are freely available at https://github.com/mapleforest/HaploMerger2/releases/.hshengf2@mail.sysu.edu.cn.Supplementary data are available at Bioinformatics online.© The Author(s) 2017. Published by Oxford University Press.
Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L.
DOI:10.1038/s41588-018-0237-2 [本文引用: 1]
Pan-genomics in the human genome era.
DOI:10.1038/s41576-020-0210-7
PMID:32034321
[本文引用: 3]
Since the early days of the genome era, the scientific community has relied on a single 'reference' genome for each species, which is used as the basis for a wide range of genetic analyses, including studies of variation within and across species. As sequencing costs have dropped, thousands of new genomes have been sequenced, and scientists have come to realize that a single reference genome is inadequate for many purposes. By sampling a diverse set of individuals, one can begin to assemble a pan-genome: a collection of all the DNA sequences that occur in a species. Here we review efforts to create pan-genomes for a range of species, from bacteria to humans, and we further consider the computational methods that have been proposed in order to capture, interpret and compare pan-genome data. As scientists continue to survey and catalogue the genomic variation across human populations and begin to assemble a human pan-genome, these efforts will increase our power to connect variation to human diversity, disease and beyond.
Pan-3D genome analysis reveals structural and functional differentiation of soybean genomes
DOI:10.1186/s13059-023-02854-8
PMID:36658660
[本文引用: 1]
High-order chromatin structure plays important roles in gene regulation. However, the diversity of the three-dimensional (3D) genome across plant accessions are seldom reported.Here, we perform the pan-3D genome analysis using Hi-C sequencing data from 27 soybean accessions and comprehensively investigate the relationships between 3D genomic variations and structural variations (SVs) as well as gene expression. We find that intersection regions between A/B compartments largely contribute to compartment divergence. Topologically associating domain (TAD) boundaries in A compartments exhibit significantly higher density compared to those in B compartments. Pan-3D genome analysis shows that core TAD boundaries have the highest transcription start site (TSS) density and lowest GC content and repeat percentage. Further investigation shows that non-long terminal repeat (non-LTR) retrotransposons play important roles in maintaining TAD boundaries, while Gypsy elements and satellite repeats are associated with private TAD boundaries. Moreover, presence and absence variation (PAV) is found to be the major contributor to 3D genome variations. Nevertheless, approximately 55% of 3D genome variations are not associated with obvious genetic variations, and half of them affect the flanking gene expression. In addition, we find that the 3D genome may also undergo selection during soybean domestication.Our study sheds light on the role of 3D genomes in plant genetic diversity and provides a valuable resource for studying gene regulation and genome evolution.© 2023. The Author(s).
Insights into the maize pan-genome and pan-transcriptome
Ten years of pan-genome analyses
DOI:10.1016/j.mib.2014.11.016
PMID:25483351
[本文引用: 2]
Next generation sequencing technologies have engendered a genome sequence data deluge in public databases. Genome analyses have transitioned from single or few genomes to hundreds to thousands of genomes. Pan-genome analyses provide a framework for estimating the genomic diversity of the dataset at hand and predicting the number of additional whole genomes sequences that would be necessary to fully characterize that diversity. We review recent implementations of the pan-genome approach, its impact and limits, and we propose possible extensions, including analyses at the whole genome multiple sequence alignment level. Copyright © 2014 Elsevier Ltd. All rights reserved.
Towards population-scale long-read sequencing
DOI:10.1038/s41576-021-00367-3
PMID:34050336
[本文引用: 1]
Long-read sequencing technologies have now reached a level of accuracy and yield that allows their application to variant detection at a scale of tens to thousands of samples. Concomitant with the development of new computational tools, the first population-scale studies involving long-read sequencing have emerged over the past 2 years and, given the continuous advancement of the field, many more are likely to follow. In this Review, we survey recent developments in population-scale long-read sequencing, highlight potential challenges of a scaled-up approach and provide guidance regarding experimental design. We provide an overview of current long-read sequencing platforms, variant calling methodologies and approaches for de novo assemblies and reference-based mapping approaches. Furthermore, we summarize strategies for variant validation, genotyping and predicting functional impact and emphasize challenges remaining in achieving long-read sequencing at a population scale.
Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure
DOI:10.1038/s41467-017-02292-8
PMID:29259172
[本文引用: 4]
While prokaryotic pan-genomes have been shown to contain many more genes than any individual organism, the prevalence and functional significance of differentially present genes in eukaryotes remains poorly understood. Whole-genome de novo assembly and annotation of 54 lines of the grass Brachypodium distachyon yield a pan-genome containing nearly twice the number of genes found in any individual genome. Genes present in all lines are enriched for essential biological functions, while genes present in only some lines are enriched for conditionally beneficial functions (e.g., defense and development), display faster evolutionary rates, lie closer to transposable elements and are less likely to be syntenic with orthologous genes in other grasses. Our data suggest that differentially present genes contribute substantially to phenotypic variation within a eukaryote species, these genes have a major influence in population genetics, and transposable elements play a key role in pan-genome evolution.
De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits
DOI:10.1038/nbt.2979
[本文引用: 8]
Wild relatives of crops are an important source of genetic diversity for agriculture, but their gene repertoire remains largely unexplored. We report the establishment and analysis of a pan-genome of Glycine soja, the wild relative of cultivated soybean Glycine max, by sequencing and de novo assembly of seven phylogenetically and geographically representative accessions. Intergenomic comparisons identified lineage-specific genes and genes with copy number variation or large-effect mutations, some of which show evidence of positive selection and may contribute to variation of agronomic traits such as biotic resistance, seed composition, flowering and maturity time, organ size and final biomass. Approximately 80% of the pan-genome was present in all seven accessions (core), whereas the rest was dispensable and exhibited greater variation than the core genome, perhaps reflecting a role in adaptation to diverse environments. This work will facilitate the harnessing of untapped genetic diversity from wild soybean for enhancement of elite cultivars.
Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza
DOI:10.1038/s41588-018-0040-0
PMID:29358651
[本文引用: 2]
The genus Oryza is a model system for the study of molecular evolution over time scales ranging from a few thousand to 15 million years. Using 13 reference genomes spanning the Oryza species tree, we show that despite few large-scale chromosomal rearrangements rapid species diversification is mirrored by lineage-specific emergence and turnover of many novel elements, including transposons, and potential new coding and noncoding genes. Our study resolves controversial areas of the Oryza phylogeny, showing a complex history of introgression among different chromosomes in the young 'AA' subclade containing the two domesticated species. This study highlights the prevalence of functionally coupled disease resistance genes and identifies many new haplotypes of potential use for future crop protection. Finally, this study marks a milestone in modern rice research with the release of a complete long-read assembly of IR 8 'Miracle Rice', which relieved famine and drove the Green Revolution in Asia 50 years ago.
The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor
DOI:10.1038/s41588-019-0410-2
PMID:31086351
[本文引用: 4]
Modern tomatoes have narrow genetic diversity limiting their improvement potential. We present a tomato pan-genome constructed using genome sequences of 725 phylogenetically and geographically representative accessions, revealing 4,873 genes absent from the reference genome. Presence/absence variation analyses reveal substantial gene loss and intense negative selection of genes and promoters during tomato domestication and improvement. Lost or negatively selected genes are enriched for important traits, especially disease resistance. We identify a rare allele in the TomLoxC promoter selected against during domestication. Quantitative trait locus mapping and analysis of transgenic plants reveal a role for TomLoxC in apocarotenoid production, which contributes to desirable tomato flavor. In orange-stage fruit, accessions harboring both the rare and common TomLoxC alleles (heterozygotes) have higher TomLoxC expression than those homozygous for either and are resurgent in modern tomatoes. The tomato pan-genome adds depth and completeness to the reference genome, and is useful for future biological discovery and breeding.
Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance
DOI:10.1038/s41477-018-0329-0
PMID:30598532
[本文引用: 5]
Domesticated plants and animals often display dramatic responses to selection, but the origins of the genetic diversity underlying these responses remain poorly understood. Despite domestication and improvement bottlenecks, the cultivated sunflower remains highly variable genetically, possibly due to hybridization with wild relatives. To characterize genetic diversity in the sunflower and to quantify contributions from wild relatives, we sequenced 287 cultivated lines, 17 Native American landraces and 189 wild accessions representing 11 compatible wild species. Cultivar sequences failing to map to the sunflower reference were assembled de novo for each genotype to determine the gene repertoire, or 'pan-genome', of the cultivated sunflower. Assembled genes were then compared to the wild species to estimate origins. Results indicate that the cultivated sunflower pan-genome comprises 61,205 genes, of which 27% vary across genotypes. Approximately 10% of the cultivated sunflower pan-genome is derived through introgression from wild sunflower species, and 1.5% of genes originated solely through introgression. Gene ontology functional analyses further indicate that genes associated with biotic resistance are over-represented among introgressed regions, an observation consistent with breeding records. Analyses of allelic variation associated with downy mildew resistance provide an example in which such introgressions have contributed to resistance to a globally challenging disease.
Genomic variation in 3,010 diverse accessions of Asian cultivated rice
The pangenome of an agronomically important crop plant Brassica oleracea
DOI:10.1038/ncomms13390
PMID:27834372
[本文引用: 6]
There is an increasing awareness that as a result of structural variation, a reference sequence representing a genome of a single individual is unable to capture all of the gene repertoire found in the species. A large number of genes affected by presence/absence and copy number variation suggest that it may contribute to phenotypic and agronomic trait diversity. Here we show by analysis of the Brassica oleracea pangenome that nearly 20% of genes are affected by presence/absence variation. Several genes displaying presence/absence variation are annotated with functions related to major agronomic traits, including disease resistance, flowering time, glucosinolate metabolism and vitamin biosynthesis.
De novo assembly and genotyping of variants using colored de Bruijn graphs
Characterizing the major structural variant alleles of the human genome
DOI:S0092-8674(18)31633-7
PMID:30661756
[本文引用: 1]
In order to provide a comprehensive resource for human structural variants (SVs), we generated long-read sequence data and analyzed SVs for fifteen human genomes. We sequence resolved 99,604 insertions, deletions, and inversions including 2,238 (1.6 Mbp) that are shared among all discovery genomes with an additional 13,053 (6.9 Mbp) present in the majority, indicating minor alleles or errors in the reference. Genotyping in 440 additional genomes confirms the most common SVs in unique euchromatin are now sequence resolved. We report a ninefold SV bias toward the last 5 Mbp of human chromosomes with nearly 55% of all VNTRs (variable number of tandem repeats) mapping to this portion of the genome. We identify SVs affecting coding and noncoding regulatory loci improving annotation and interpretation of functional variation. These data provide the framework to construct a canonical human reference and a resource for developing advanced representations capable of capturing allelic diversity.Copyright © 2018 Elsevier Inc. All rights reserved.
GraphTyper 2 enables population-scale genotyping of structural variation using pangenome graphs
DOI:10.1038/s41467-019-13341-9
PMID:31776332
[本文引用: 2]
Analysis of sequence diversity in the human genome is fundamental for genetic studies. Structural variants (SVs) are frequently omitted in sequence analysis studies, although each has a relatively large impact on the genome. Here, we present GraphTyper2, which uses pangenome graphs to genotype SVs and small variants using short-reads. Comparison to the syndip benchmark dataset shows that our SV genotyping is sensitive and variant segregation in families demonstrates the accuracy of our approach. We demonstrate that incorporating public assembly data into our pipeline greatly improves sensitivity, particularly for large insertions. We validate 6,812 SVs on average per genome using long-read data of 41 Icelanders. We show that GraphTyper2 can simultaneously genotype tens of thousands of whole-genomes by characterizing 60 million small variants and half a million SVs in 49,962 Icelanders, including 80 thousand SVs with high-confidence.
Variation graph toolkit improves read mapping by representing genetic variation in the reference
DOI:10.1038/nbt.4227
PMID:30125266
[本文引用: 2]
Reference genomes guide our interpretation of DNA sequence data. However, conventional linear references represent only one version of each locus, ignoring variation in the population. Poor representation of an individual's genome sequence impacts read mapping and introduces bias. Variation graphs are bidirected DNA sequence graphs that compactly represent genetic variation across a population, including large-scale structural variation such as inversions and duplications. Previous graph genome software implementations have been limited by scalability or topological constraints. Here we present vg, a toolkit of computational methods for creating, manipulating, and using these structures as references at the scale of the human genome. vg provides an efficient approach to mapping reads onto arbitrary variation graphs using generalized compressed suffix arrays, with improved accuracy over alignment to a linear reference, and effectively removing reference bias. These capabilities make using variation graphs as references for DNA sequencing practical at a gigabase scale, or at the topological complexity of de novo assemblies.
SplitMEM: a graphical algorithm for pan-genome analysis with suffix skips
DOI:10.1093/bioinformatics/btu756
PMID:25398610
[本文引用: 1]
Genomics is expanding from a single reference per species paradigm into a more comprehensive pan-genome approach that analyzes multiple individuals together. A compressed de Bruijn graph is a sophisticated data structure for representing the genomes of entire populations. It robustly encodes shared segments, simple single-nucleotide polymorphisms and complex structural variations far beyond what can be represented in a collection of linear sequences alone.We explore deep topological relationships between suffix trees and compressed de Bruijn graphs and introduce an algorithm, splitMEM, that directly constructs the compressed de Bruijn graph in time and space linear to the total number of genomes for a given maximum genome size. We introduce suffix skips to traverse several suffix links simultaneously and use them to efficiently decompose maximal exact matches into graph nodes. We demonstrate the utility of splitMEM by analyzing the nine-strain pan-genome of Bacillus anthracis and up to 62 strains of Escherichia coli, revealing their core-genome properties.© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
PanGP: a tool for quickly analyzing bacterial pan-genome profile
DOI:10.1093/bioinformatics/btu017
PMID:24420766
[本文引用: 1]
Pan-genome analyses have shed light on the dynamics and evolution of bacterial genome from the point of population. The explosive growth of bacterial genome sequence also brought an extremely big challenge to pan-genome profile analysis. We developed a tool, named PanGP, to complete pan-genome profile analysis for large-scale strains efficiently. PanGP has integrated two sampling algorithms, totally random (TR) and distance guide (DG). The DG algorithm drew sample strain combinations on the basis of genome diversity of bacterial population. The performance of these two algorithms have been evaluated on four bacteria populations with strain numbers varying from 30 to 200, and the DG algorithm exhibited overwhelming advantage on accuracy and stability than the TR algorithm.
Pangenomics enables genotyping of known structural variants in 5202 diverse genomes
DOI:10.1126/science.abg8871
URL
[本文引用: 1]
\n Genomes within a species often have a core, conserved component, as well as a variable set of genetic material among individuals or populations that is referred to as a “pangenome.” Inference of the relationships between pangenomes sequenced with short-read technology is often done computationally by mapping the sequences to a reference genome. The computational method affects genome assembly and comparisons, especially in cases of structural variants that are longer than an average sequenced region, for highly polymorphic loci, and for cross-species analyses. Siren\n et al\n. present a bioinformatic method called Giraffe, which improves mapping pangenomes in polymorphic regions of the genome containing single nucleotide polymorphisms and structural variants with standard computational resources, making large-scale genomic analyses more accessible. —LMZ\n
ODGI: understanding pangenome graphs
DOI:10.1093/bioinformatics/btac308
PMID:35552372
[本文引用: 1]
Pangenome graphs provide a complete representation of the mutual alignment of collections of genomes. These models offer the opportunity to study the entire genomic diversity of a population, including structurally complex regions. Nevertheless, analyzing hundreds of gigabase-scale genomes using pangenome graphs is difficult as it is not well-supported by existing tools. Hence, fast and versatile software is required to ask advanced questions to such data in an efficient way.We wrote ODGI, a novel suite of tools that implements scalable algorithms and has an efficient in-memory representation of DNA pangenome graphs in the form of variation graphs. ODGI supports pre-built graphs in the Graphical Fragment Assembly format. ODGI includes tools for detecting complex regions, extracting pangenomic loci, removing artifacts, exploratory analysis, manipulation, validation, and visualization. Its fast parallel execution facilitates routine pangenomic tasks, as well as pipelines that can quickly answer complex biological questions of gigabase-scale pangenome graphs.ODGI is published as free software under the MIT open source license. Source code can be downloaded from https://github.com/pangenome/odgi and documentation is available at https://odgi.readthedocs.io. ODGI can be installed via Bioconda https://bioconda.github.io/recipes/odgi/README.html or GNU Guix https://github.com/pangenome/odgi/blob/master/guix.scm.© The Author(s) 2022. Published by Oxford University Press.
Building pangenome graphs
Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype
DOI:10.1038/s41587-019-0201-4
PMID:31375807
[本文引用: 1]
The human reference genome represents only a small number of individuals, which limits its usefulness for genotyping. We present a method named HISAT2 (hierarchical indexing for spliced alignment of transcripts 2) that can align both DNA and RNA sequences using a graph Ferragina Manzini index. We use HISAT2 to represent and search an expanded model of the human reference genome in which over 14.5 million genomic variants in combination with haplotypes are incorporated into the data structure used for searching and alignment. We benchmark HISAT2 using simulated and real datasets to demonstrate that our strategy of representing a population of genomes, together with a fast, memory-efficient search algorithm, provides more detailed and accurate variant analyses than other methods. We apply HISAT2 for HLA typing and DNA fingerprinting; both applications form part of the HISAT-genotype software that enables analysis of haplotype-resolved genes or genomic regions. HISAT-genotype outperforms other computational methods and matches or exceeds the performance of laboratory-based assays.
Multiple reference genomes and transcriptomes for Arabidopsis thaliana
DOI:10.1038/nature10414 [本文引用: 2]
Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations
DOI:10.1016/j.cell.2021.04.046
PMID:34051138
[本文引用: 3]
Structural variations (SVs) and gene copy number variations (gCNVs) have contributed to crop evolution, domestication, and improvement. Here, we assembled 31 high-quality genomes of genetically diverse rice accessions. Coupling with two existing assemblies, we developed pan-genome-scale genomic resources including a graph-based genome, providing access to rice genomic variations. Specifically, we discovered 171,072 SVs and 25,549 gCNVs and used an Oryza glaberrima assembly to infer the derived states of SVs in the Oryza sativa population. Our analyses of SV formation mechanisms, impacts on gene expression, and distributions among subpopulations illustrate the utility of these resources for understanding how SVs and gCNVs shaped rice environmental adaptation and domestication. Our graph-based genome enabled genome-wide association study (GWAS)-based identification of phenotype-associated genetic variations undetectable when using only SNPs and a single reference assembly. Our work provides rich population-scale resources paired with easy-to-access tools to facilitate rice breeding as well as plant functional genomics and evolutionary biology research.Copyright © 2021 Elsevier Inc. All rights reserved.
Graph pangenome captures missing heritability and empowers tomato breeding
DOI:10.1038/s41586-022-04808-9
[本文引用: 3]
Missing heritability in genome-wide association studies defines a major problem in genetic analyses of complex biological traits1,2. The solution to this problem is to identify all causal genetic variants and to measure their individual contributions3,4. Here we report a graph pangenome of tomato constructed by precisely cataloguing more than 19 million variants from 838 genomes, including 32 new reference-level genome assemblies. This graph pangenome was used for genome-wide association study analyses and heritability estimation of 20,323 gene-expression and metabolite traits. The average estimated trait heritability is 0.41 compared with 0.33 when using the single linear reference genome. This 24% increase in estimated heritability is largely due to resolving incomplete linkage disequilibrium through the inclusion of additional causal structural variants identified using the graph pangenome. Moreover, by resolving allelic and locus heterogeneity, structural variants improve the power to identify genetic factors underlying agronomically important traits leading to, for example, the identification of two new genes potentially contributing to soluble solid content. The newly identified structural variants will facilitate genetic improvement of tomato through both marker-assisted selection and genomic selection. Our study advances the understanding of the heritability of complex traits and demonstrates the power of the graph pangenome in crop breeding.
Pangenome analysis provides insight into the evolution of the orange subfamily and a key gene for citric acid accumulation in citrus fruits
DOI:10.1038/s41588-023-01516-6
PMID:37783780
[本文引用: 2]
The orange subfamily (Aurantioideae) contains several Citrus species cultivated worldwide, such as sweet orange and lemon. The origin of Citrus species has long been debated and less is known about the Aurantioideae. Here, we compiled the genome sequences of 314 accessions, de novo assembled the genomes of 12 species and constructed a graph-based pangenome for Aurantioideae. Our analysis indicates that the ancient Indian Plate is the ancestral area for Citrus-related genera and that South Central China is the primary center of origin of the Citrus genus. We found substantial variations in the sequence and expression of the PH4 gene in Citrus relative to Citrus-related genera. Gene editing and biochemical experiments demonstrate a central role for PH4 in the accumulation of citric acid in citrus fruits. This study provides insights into the origin and evolution of the orange subfamily and a regulatory mechanism underpinning the evolution of fruit taste.© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Structural variation (SV)-based pan-genome and GWAS reveal the impacts of SVs on the speciation and diversification of allotetraploid cottons
DOI:10.1016/j.molp.2023.02.004
PMID:36760124
[本文引用: 1]
Structural variations (SVs) have long been described as involved in the origin, adaption, and domestication of species. However, the genetic and genomic mechanisms of that involvement are poorly understood. Here, we assembled a high-quality genome of Gossypium barbadense acc. Tanguis, a landrace which is closely connected to the formation of extra-long‒staple (ELS) cultivated cotton. A SV-based pan-genome (Pan-SV) was constructed using a total of 182,593 non-redundant SVs, including 2,236 inversions, 97,398 insertions, and 82,959 deletions from 11 assembled genomes of allopolyploid cotton. The utility of this Pan-SV was then demonstrated through population structure analysis and genome-wide association studies (GWAS). Using segregation mapping populations produced through crossing ELS cotton and the landrace along with a SV-based GWAS, certain SVs responsible for speciation, domestication, and improvement in tetraploid cottons were identified. Importantly, some of the SVs presently identified as relating to yield and fiber quality improvement had not been identified in previous SNP-based GWAS. In particular, a 9-bp indel was found to associate with elimination of the interspecific reproductive isolation between G. hirsutum and G. barbadense. This study provides insights into genome-wide, gene-scale SVs linked to important agronomic traits in a major crop species and highlights the importance of SVs during the speciation, domestication, and improvement of cultivated crop species.Copyright © 2023 The Author. Published by Elsevier Inc. All rights reserved.
Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber
DOI:10.1038/s41467-022-28362-0
PMID:35115520
[本文引用: 2]
Structural variants (SVs) represent a major source of genetic diversity and are related to numerous agronomic traits and evolutionary events; however, their comprehensive identification and characterization in cucumber (Cucumis sativus L.) have been hindered by the lack of a high-quality pan-genome. Here, we report a graph-based cucumber pan-genome by analyzing twelve chromosome-scale genome assemblies. Genotyping of seven large chromosomal rearrangements based on the pan-genome provides useful information for use of wild accessions in breeding and genetic studies. A total of ~4.3 million genetic variants including 56,214 SVs are identified leveraging the chromosome-level assemblies. The pan-genome graph integrating both variant information and reference genome sequences aids the identification of SVs associated with agronomic traits, including warty fruits, flowering times and root growth, and enhances the understanding of cucumber trait evolution. The graph-based cucumber pan-genome and the identified genetic variants provide rich resources for future biological research and genomics-assisted breeding.© 2022. The Author(s).
Pan-genome of wild and cultivated soybeans
DOI:S0092-8674(20)30618-8
PMID:32553274
[本文引用: 11]
Soybean is one of the most important vegetable oil and protein feed crops. To capture the entire genomic diversity, it is needed to construct a complete high-quality pan-genome from diverse soybean accessions. In this study, we performed individual de novo genome assemblies for 26 representative soybeans that were selected from 2,898 deeply sequenced accessions. Using these assembled genomes together with three previously reported genomes, we constructed a graph-based genome and performed pan-genome analysis, which identified numerous genetic variations that cannot be detected by direct mapping of short sequence reads onto a single reference genome. The structural variations from the 2,898 accessions that were genotyped based on the graph-based genome and the RNA sequencing (RNA-seq) data from the representative 26 accessions helped to link genetic variations to candidate genes that are responsible for important traits. This pan-genome resource will promote evolutionary and functional genomics studies in soybean.Copyright © 2020 Elsevier Inc. All rights reserved.
A graph-based genome and pan-genome variation of the model plant Setaria
DOI:10.1038/s41588-023-01423-w
PMID:37291196
[本文引用: 6]
Setaria italica (foxtail millet), a founder crop of East Asian agriculture, is a model plant for C4 photosynthesis and developing approaches to adaptive breeding across multiple climates. Here we established the Setaria pan-genome by assembling 110 representative genomes from a worldwide collection. The pan-genome is composed of 73,528 gene families, of which 23.8%, 42.9%, 29.4% and 3.9% are core, soft core, dispensable and private genes, respectively; 202,884 nonredundant structural variants were also detected. The characterization of pan-genomic variants suggests their importance during foxtail millet domestication and improvement, as exemplified by the identification of the yield gene SiGW3, where a 366-bp presence/absence promoter variant accompanies gene expression variation. We developed a graph-based genome and performed large-scale genetic studies for 68 traits across 13 environments, identifying potential genes for millet improvement at different geographic sites. These can be used in marker-assisted breeding, genomic selection and genome editing to accelerate crop improvement under different climatic conditions.© 2023. The Author(s).
Gene mining and genomics-assisted breeding empowered by the pangenome of tea plant Camellia sinensis
DOI:10.1038/s41477-023-01565-z
PMID:38012346
[本文引用: 3]
Tea is one of the world's oldest crops and is cultivated to produce beverages with various flavours. Despite advances in sequencing technologies, the genetic mechanisms underlying key agronomic traits of tea remain unclear. In this study, we present a high-quality pangenome of 22 elite cultivars, representing broad genetic diversity in the species. Our analysis reveals that a recent long terminal repeat burst contributed nearly 20% of gene copies, introducing functional genetic variants that affect phenotypes such as leaf colour. Our graphical pangenome improves the efficiency of genome-wide association studies and allows the identification of key genes controlling bud flush timing. We also identified strong correlations between allelic variants and flavour-related chemistries. These findings deepen our understanding of the genetic basis of tea quality and provide valuable genomic resources to facilitate its genomics-assisted breeding.© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice
DOI:10.1038/s41588-018-0041-z
PMID:29335547
[本文引用: 2]
The rich genetic diversity in Oryza sativa and Oryza rufipogon serves as the main sources in rice breeding. Large-scale resequencing has been undertaken to discover allelic variants in rice, but much of the information for genetic variation is often lost by direct mapping of short sequence reads onto the O. sativa japonica Nipponbare reference genome. Here we constructed a pan-genome dataset of the O. sativa-O. rufipogon species complex through deep sequencing and de novo assembly of 66 divergent accessions. Intergenomic comparisons identified 23 million sequence variants in the rice genome. This catalog of sequence variations includes many known quantitative trait nucleotides and will be helpful in pinpointing new causal variants that underlie complex traits. In particular, we systemically investigated the whole set of coding genes using this pan-genome data, which revealed extensive presence and absence of variation among rice accessions. This pan-genome resource will further promote evolutionary and functional studies in rice.
Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus
DOI:10.1038/s41477-019-0577-7
[本文引用: 2]
Rapeseed (Brassica napus) is the second most important oilseed crop in the world but the genetic diversity underlying its massive phenotypic variations remains largely unexplored. Here, we report the sequencing, de novo assembly and annotation of eight B. napus accessions. Using pan-genome comparative analysis, millions of small variations and 77.2–149.6 megabase presence and absence variations (PAVs) were identified. More than 9.4% of the genes contained large-effect mutations or structural variations. PAV-based genome-wide association study (PAV-GWAS) directly identified causal structural variations for silique length, seed weight and flowering time in a nested association mapping population with ZS11 (reference line) as the donor, which were not detected by single-nucleotide polymorphisms-based GWAS (SNP-GWAS), demonstrating that PAV-GWAS was complementary to SNP-GWAS in identifying associations to traits. Further analysis showed that PAVs in three FLOWERING LOCUS C genes were closely related to flowering time and ecotype differentiation. This study provides resources to support a better understanding of the genome architecture and acceleration of the genetic improvement of B. napus.
A super pan-genomic landscape of rice
DOI:10.1038/s41422-022-00685-z
PMID:35821092
[本文引用: 1]
Pan-genomes from large natural populations can capture genetic diversity and reveal genomic complexity. Using de novo long-read assembly, we generated a graph-based super pan-genome of rice consisting of a 251-accession panel comprising both cultivated and wild species of Asian and African rice. Our pan-genome reveals extensive structural variations (SVs) and gene presence/absence variations. Additionally, our pan-genome enables the accurate identification of nucleotide-binding leucine-rich repeat genes and characterization of their inter- and intraspecific diversity. Moreover, we uncovered grain weight-associated SVs which specify traits by affecting the expression of their nearby genes. We characterized genetic variants associated with submergence tolerance, seed shattering and plant architecture and found independent selection for a common set of genes that drove adaptation and domestication in Asian and African rice. This super pan-genome facilitates pinpointing of lineage-specific haplotypes for trait-associated genes and provides insights into the evolutionary events that have shaped the genomic architecture of various rice species.© 2022. The Author(s).
Major impacts of widespread structural variation on gene expression and crop improvement in tomato
DOI:S0092-8674(20)30616-4
PMID:32553272
[本文引用: 2]
Structural variants (SVs) underlie important crop improvement and domestication traits. However, resolving the extent, diversity, and quantitative impact of SVs has been challenging. We used long-read nanopore sequencing to capture 238,490 SVs in 100 diverse tomato lines. This panSV genome, along with 14 new reference assemblies, revealed large-scale intermixing of diverse genotypes, as well as thousands of SVs intersecting genes and cis-regulatory regions. Hundreds of SV-gene pairs exhibit subtle and significant expression changes, which could broadly influence quantitative trait variation. By combining quantitative genetics with genome editing, we show how multiple SVs that changed gene dosage and expression levels modified fruit flavor, size, and production. In the last example, higher order epistasis among four SVs affecting three related transcription factors allowed introduction of an important harvesting trait in modern tomato. Our findings highlight the underexplored role of SVs in genotype-to-phenotype relationships and their widespread importance and utility in crop improvement.Copyright © 2020 Elsevier Inc. All rights reserved.
Pangenome analysis reveals genomic variations associated with domestication traits in broomcorn millet
DOI:10.1038/s41588-023-01571-z
[本文引用: 2]
Broomcorn millet (Panicum miliaceum L.) is an orphan crop with the potential to improve cereal production and quality, and ensure food security. Here we present the genetic variations, population structure and diversity of a diverse worldwide collection of 516 broomcorn millet genomes. Population analysis indicated that the domesticated broomcorn millet originated from its wild progenitor in China. We then constructed a graph-based pangenome of broomcorn millet based on long-read de novo genome assemblies of 32 representative accessions. Our analysis revealed that the structural variations were highly associated with transposable elements, which influenced gene expression when located in the coding or regulatory regions. We also identified 139 loci associated with 31 key domestication and agronomic traits, including candidate genes and superior haplotypes, such as LG1, for panicle architecture. Thus, the study’s findings provide foundational resources for developing genomics-assisted breeding programs in broomcorn millet.
Soybean seed lustre phenotype and surface protein cosegregate and map to linkage group E
DOI:10.1139/g03-047
PMID:12897873
[本文引用: 1]
Soybean (Glycine max (L.) Merr.) seeds vary in their surface properties. The lustre, or glossiness, of seeds has been classified into several different phenotypes. Soybean seeds that have a dull lustre or moderate bloom (B) may also have abundant seed surface protein, namely, an abundance of the hydrophobic protein from soybean (HPS). The seed surface protein HPS is an allergen (Gly m 1) that causes asthma in persons allergic to soybean dust. In this study, seed lustre and surface protein content are compared among 71 different soybean cultivars and lines. Dull-seeded phenotypes usually possessed abundant surface protein in comparison to shiny-seeded types, although exceptions were observed. An F2 population of 82 individuals from a cross of OX281 (dull lustre, abundant HPS) and Mukden (shiny lustre, trace amounts of HPS) provided a basis for inheritance studies and genetic mapping analysis. Results indicate that dull seed lustre (B) and surface protein (Hps) loci are dominant Mendelian traits that cosegregate and map to soybean linkage group E. Molecular markers were used to construct a genetic map of 28 cM encompassing B and Hps. Two different molecular markers cosegregated with each of the loci. This study provides additional evidence that Hps may play a role in the adhesion of endocarp tissues to the seed, and offers new methods of selection for seed lustre and surface protein composition in soybean.
Parallel evolution of chimeric fusion genes
To understand how novel functions arise, we must identify common patterns and mechanisms shaping the evolution of new genes. Here, we take advantage of data from three Drosophila genes, jingwei, Adh-Finnegan, and Adh-Twain, to find evolutionary patterns and mechanisms governing the evolution of new genes. All three of these genes are independently derived from Adh, which enabled us to use the extensive literature on Adh in Drosophila to guide our analyses. We discovered a fundamental similarity in the temporal, spatial, and types of amino acid changes that occurred. All three genes underwent rapid adaptive amino acid evolution shortly after they were formed, followed by later quiescence and functional constraint. These genes also show striking parallels in which amino acids change in the Adh region. We showed that these early changes tend to occur at amino acid residues that seldom, if ever, evolve in Drosophila Adh. Changes at these slowly evolving sites are usually associated with loss of function or hypomorphic mutations in Drosophila melanogaster. Our data indicate that shifting away from ancestral functions may be a critical step early in the evolution of chimeric fusion genes. We suggest that the patterns we observed are both general and predictive.
Map-based cloning of the gene associated with the soybean maturity locus E3
DOI:10.1534/genetics.108.098772
PMID:19474204
[本文引用: 1]
Photosensitivity plays an essential role in the response of plants to their changing environments throughout their life cycle. In soybean [Glycine max (L.) Merrill], several associations between photosensitivity and maturity loci are known, but only limited information at the molecular level is available. The FT3 locus is one of the quantitative trait loci (QTL) for flowering time that corresponds to the maturity locus E3. To identify the gene responsible for this QTL, a map-based cloning strategy was undertaken. One phytochrome A gene (GmPhyA3) was considered a strong candidate for the FT3 locus. Allelism tests and gene sequence comparisons showed that alleles of Misuzudaizu (FT3/FT3; JP28856) and Harosoy (E3/E3; PI548573) were identical. The GmPhyA3 alleles of Moshidou Gong 503 (ft3/ft3; JP27603) and L62-667 (e3/e3; PI547716) showed weak or complete loss of function, respectively. High red/far-red (R/FR) long-day conditions enhanced the effects of the E3/FT3 alleles in various genetic backgrounds. Moreover, a mutant line harboring the nonfunctional GmPhyA3 flowered earlier than the original Bay (E3/E3; PI553043) under similar conditions. These results suggest that the variation in phytochrome A may contribute to the complex systems of soybean flowering response and geographic adaptation.
Natural variation in the genes responsible for maturity loci E1, E2, E3 and E4 in soybean
DOI:10.1093/aob/mct269 URL [本文引用: 1]
Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection
Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield
Soybean is a major legume crop originating in temperate regions, and photoperiod responsiveness is a key factor in its latitudinal adaptation. Varieties from temperate regions introduced to lower latitudes mature early and have extremely low grain yields. Introduction of the long-juvenile (LJ) trait extends the vegetative phase and improves yield under short-day conditions, thereby enabling expansion of cultivation in tropical regions. Here we report the cloning and characterization of J, the major classical locus conferring the LJ trait, and identify J as the ortholog of Arabidopsis thaliana EARLY FLOWERING 3 (ELF3). J depends genetically on the legume-specific flowering repressor E1, and J protein physically associates with the E1 promoter to downregulate its transcription, relieving repression of two important FLOWERING LOCUS T (FT) genes and promoting flowering under short days. Our findings identify an important new component in flowering-time control in soybean and provide new insight into soybean adaptation to tropical regions.
Comprehensive description of genomewide nucleotide and structural variation in short-season soya bean
DOI:10.1111/pbi.12825
PMID:28869792
[本文引用: 1]
Next-generation sequencing (NGS) and bioinformatics tools have greatly facilitated the characterization of nucleotide variation; nonetheless, an exhaustive description of both SNP haplotype diversity and of structural variation remains elusive in most species. In this study, we sequenced a representative set of 102 short-season soya beans and achieved an extensive coverage of both nucleotide diversity and structural variation (SV). We called close to 5M sequence variants (SNPs, MNPs and indels) and noticed that the number of unique haplotypes had plateaued within this set of germplasm (1.7M tag SNPs). This data set proved highly accurate (98.6%) based on a comparison of called genotypes at loci shared with a SNP array. We used this catalogue of SNPs as a reference panel to impute missing genotypes at untyped loci in data sets derived from lower density genotyping tools (150 K GBS-derived SNPs/530 samples). After imputation, 96.4% of the missing genotypes imputed in this fashion proved to be accurate. Using a combination of three bioinformatics pipelines, we uncovered ~92 K SVs (deletions, insertions, inversions, duplications, CNVs and translocations) and estimated that over 90% of these were accurate. Finally, we noticed that the duplication of certain genomic regions explained much of the residual heterozygosity at SNP loci in otherwise highly inbred soya bean accessions. This is the first time that a comprehensive description of both SNP haplotype diversity and SV has been achieved within a regionally relevant subset of a major crop.© 2017 The Authors. Plant Biotechnology Journal published by Society for Experimental Biology and The Association of Applied Biologists and John Wiley & Sons Ltd.
Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean
DOI:10.1038/nbt.3096
PMID:25643055
[本文引用: 2]
Understanding soybean (Glycine max) domestication and improvement at a genetic level is important to inform future efforts to further improve a crop that provides the world's main source of oilseed. We detect 230 selective sweeps and 162 selected copy number variants by analysis of 302 resequenced wild, landrace and improved soybean accessions at >11× depth. A genome-wide association study using these new sequences reveals associations between 10 selected regions and 9 domestication or improvement traits, and identifies 13 previously uncharacterized loci for agronomic traits including oil content, plant height and pubescence form. Combined with previous quantitative trait loci (QTL) information, we find that, of the 230 selected regions, 96 correlate with reported oil QTLs and 21 contain fatty acid biosynthesis genes. Moreover, we observe that some traits and loci are associated with geographical regions, which shows that soybean populations are structured geographically. This study provides resources for genomics-enabled improvements in soybean breeding.
Inheritance of cotyledon, seed-coat, hilum and pubescence colors in soy-beans
DOI:10.1093/genetics/6.6.487 PMID:17245974 [本文引用: 1]
Tissue- specific gene silencing mediated by a naturally occurring chalcone synthase gene cluster in Glycine max
DOI:10.1105/tpc.021352
URL
[本文引用: 1]
Chalcone synthase, a key regulatory enzyme in the flavonoid pathway, constitutes an eight-member gene family in Glycine max (soybean). Three of the chalcone synthase (CHS) gene family members are arranged as inverted repeats in a 10-kb region, corresponding to the I locus (inhibitor). Spontaneous mutations of a dominant allele (I or ii) to a recessive allele (i) have been shown to delete promoter sequences, paradoxically increasing total CHS transcript levels and resulting in black seed coats. However, it is not known which of the gene family members contribute toward pigmentation and how this locus affects CHS expression in other tissues. We investigated the unusual nature of the I locus using four pairs of isogenic lines differing with respect to alleles of the I locus. RNA gel blots using a generic open reading frame CHS probe detected similar CHS transcript levels in stems, roots, leaves, young pods, and cotyledons of the yellow and black isolines but not in the seed coats, which is consistent with the dominant I and ii alleles mediating CHS gene silencing in a tissue-specific manner. Using real-time RT-PCR, a variable pattern of expression of CHS genes in different tissues was demonstrated. However, increase in pigmentation in the black seed coats was associated with release of the silencing effect specifically on CHS7/CHS8, which occurred at all stages of seed coat development. These expression changes were linked to structural changes taking place at the I locus, shown to encompass a much wider region of at least 27 kb, comprising two identical 10.91-kb stretches of CHS gene duplications. The suppressive effect of this 27-kb I locus in a specific tissue of the G. max plant represents a unique endogenous gene silencing mechanism.
Endogenous, tissue-specific short interfering RNAs silence the chalcone synthase gene family in Glycine max seed coats
DOI:10.1105/tpc.109.069856
URL
[本文引用: 1]
Two dominant alleles of the I locus in Glycine max silence nine chalcone synthase (CHS) genes to inhibit function of the flavonoid pathway in the seed coat. We describe here the intricacies of this naturally occurring silencing mechanism based on results from small RNA gel blots and high-throughput sequencing of small RNA populations. The two dominant alleles of the I locus encompass a 27-kb region containing two perfectly repeated and inverted clusters of three chalcone synthase genes (CHS1, CHS3, and CHS4). This structure silences the expression of all CHS genes, including CHS7 and CHS8, located on other chromosomes. The CHS short interfering RNAs (siRNAs) sequenced support a mechanism by which RNAs transcribed from the CHS inverted repeat form aberrant double-stranded RNAs that become substrates for dicer-like ribonuclease. The resulting primary siRNAs become guides that target the mRNAs of the nonlinked, highly expressed CHS7 and CHS8 genes, followed by subsequent amplification of CHS7 and CHS8 secondary siRNAs by RNA-dependent RNA polymerase. Most remarkably, this silencing mechanism occurs only in one tissue, the seed coat, as shown by the lack of CHS siRNAs in cotyledons and vegetative tissues. Thus, production of the trigger double-stranded RNA that initiates the process occurs in a specific tissue and represents an example of naturally occurring inhibition of a metabolic pathway by siRNAs in one tissue while allowing expression of the pathway and synthesis of valuable secondary metabolites in all other organs/tissues of the plant.
Chalcone synthase mRNA and activity are reduced in yellow soybean seed coats with dominant I alleles
DOI:10.1104/pp.105.2.739
PMID:8066134
[本文引用: 1]
The seed of all wild Glycine accessions have black or brown pigments because of the homozygous recessive i allele in combination with alleles at the R and T loci. In contrast, nearly all commercial soybean (Glycine max) varieties are yellow due to the presence of a dominant allele of the I locus (either I or i) that inhibits pigmentation in the seed coats. Spontaneous mutations to the recessive i allele occur in these varieties and result in pigmented seed coats. We have isolated a clone for a soybean dihydroflavonol reductase (DFR) gene using polymerase chain reaction. We examined expression of DFR and two other genes of the flavonoid pathway during soybean seed coat development in a series of near-isogenic isolines that vary in pigmentation as specified by combinations of alleles of the I, R, and T loci. The expression of phenylalanine ammonia-lyase and DFR mRNAs was similar in all of the gene combinations at each stage of seed coat development. In contrast, chalcone synthase (CHS) mRNA was barely detectable at all stages of development in seed coats that carry the dominant I allele that results in yellow seed coats. CHS activity in yellow seed coats (I) was also 7- to 10-fold less than in the pigmented seed coats that have the homozygous recessive i allele. It appears that the dominant I allele results in reduction of CHS mRNA, leading to reduction of CHS activity as the basis for inhibition of anthocyanin and proanthocyanin synthesis in soybean seed coats. A further connection between CHS and the I locus is indicated by the occurrence of multiple restriction site polymorphisms in genomic DNA blots of the CHS gene family in near-isogenic lines containing alleles of the I locus.
A reference-grade wild soybean genome
DOI:10.1038/s41467-019-09142-9
PMID:30872580
[本文引用: 1]
Efficient crop improvement depends on the application of accurate genetic information contained in diverse germplasm resources. Here we report a reference-grade genome of wild soybean accession W05, with a final assembled genome size of 1013.2 Mb and a contig N50 of 3.3 Mb. The analytical power of the W05 genome is demonstrated by several examples. First, we identify an inversion at the locus determining seed coat color during domestication. Second, a translocation event between chromosomes 11 and 13 of some genotypes is shown to interfere with the assignment of QTLs. Third, we find a region containing copy number variations of the Kunitz trypsin inhibitor (KTI) genes. Such findings illustrate the power of this assembly in the analysis of large structural variations in soybean germplasm collections. The wild soybean genome assembly has wide applications in comparative genomic and evolutionary studies, as well as in crop breeding and improvement programs.
Mapping genetic loci for iron deficiency chlorosis in soybean
DOI:10.1023/A:1009637320805 URL [本文引用: 1]
A unified classification system for eukaryotic transposable elements
DOI:10.1038/nrg2165
PMID:17984973
[本文引用: 1]
Our knowledge of the structure and composition of genomes is rapidly progressing in pace with their sequencing. The emerging data show that a significant portion of eukaryotic genomes is composed of transposable elements (TEs). Given the abundance and diversity of TEs and the speed at which large quantities of sequence data are emerging, identification and annotation of TEs presents a significant challenge. Here we propose the first unified hierarchical classification system, designed on the basis of the transposition mechanism, sequence similarities and structural relationships, that can be easily applied by non-experts. The system and nomenclature is kept up to date at the WikiPoson web site.
Phylogenomics of the genus Glycine sheds light on polyploid evolution and life-strategy transition
DOI:10.1038/s41477-022-01102-4 [本文引用: 4]
The wondrous cycles of polyploidy in plants
DOI:10.3732/ajb.1500320 PMID:26451037 [本文引用: 1]
Patterns and consequences of subgenome differentiation provide insights into the nature of paleopolyploidy in plants
DOI:10.1105/tpc.17.00595 URL [本文引用: 1]
Goodbye reference, hello genome graphs
DOI:10.1038/s41587-019-0199-7 PMID:31375808 [本文引用: 1]
Plant pan-genomes are the new reference
DOI:10.1038/s41477-020-0733-0
PMID:32690893
[本文引用: 1]
Recent years have seen a surge in plant genome sequencing projects and the comparison of multiple related individuals. The high degree of genomic variation observed led to the realization that single reference genomes do not represent the diversity within a species, and led to the expansion of the pan-genome concept. Pan-genomes represent the genomic diversity of a species and includes core genes, found in all individuals, as well as variable genes, which are absent in some individuals. Variable gene annotations often show similarities across plant species, with genes for biotic and abiotic stress commonly enriched within variable gene groups. Here we review the growth of pan-genomics in plants, explore the origins of gene presence and absence variation, and show how pan-genomes can support plant breeding and evolution studies.
Genomic analysis in the age of human genome sequencing
DOI:S0092-8674(19)30215-6
PMID:30901550
[本文引用: 1]
Affordable genome sequencing technologies promise to revolutionize the field of human genetics by enabling comprehensive studies that interrogate all classes of genome variation, genome-wide, across the entire allele frequency spectrum. Ongoing projects worldwide are sequencing many thousands-and soon millions-of human genomes as part of various gene mapping studies, biobanking efforts, and clinical programs. However, while genome sequencing data production has become routine, genome analysis and interpretation remain challenging endeavors with many limitations and caveats. Here, we review the current state of technologies for genetic variant discovery, genotyping, and functional interpretation and discuss the prospects for future advances. We focus on germline variants discovered by whole-genome sequencing, genome-wide functional genomic approaches for predicting and measuring variant functional effects, and implications for studies of common and rare human disease.Copyright © 2019 Elsevier Inc. All rights reserved.
The integrated genomics of crop domestication and breeding
DOI:10.1016/j.cell.2022.04.036
PMID:35643084
[本文引用: 1]
As a major event in human civilization, wild plants were successfully domesticated to be crops, largely owing to continuing artificial selection. Here, we summarize new discoveries made during the past decade in crop domestication and breeding. The construction of crop genome maps and the functional characterization of numerous trait genes provide foundational information. Approaches to read, interpret, and write complex genetic information are being leveraged in many plants for highly efficient de novo or re-domestication. Understanding the underlying mechanisms of crop microevolution and applying the knowledge to agricultural productions will give possible solutions for future challenges in food security.Copyright © 2022 Elsevier Inc. All rights reserved.
Plant pan-genomics and its applications
DOI:10.1016/j.molp.2022.12.009 URL [本文引用: 1]
Plant pan-genomics comes of age.
DOI:10.1146/annurev-arplant-080720-105454
PMID:33848428
[本文引用: 1]
A pan-genome is the nonredundant collection of genes and/or DNA sequences in a species. Numerous studies have shown that plant pan-genomes are typically much larger than the genome of any individual and that a sizable fraction of the genes in any individual are present in only some genomes. The construction and interpretation of plant pan-genomes are challenging due to the large size and repetitive content of plant genomes. Most pan-genomes are largely focused on nontransposable element protein coding genes because they are more easily analyzed and defined than noncoding and repetitive sequences. Nevertheless, noncoding and repetitive DNA play important roles in determining the phenotype and genome evolution. Fortunately, it is now feasible to make multiple high-quality genomes that can be used to construct high-resolution pan-genomes that capture all the variation. However, assembling, displaying, and interacting with such high-resolution pan-genomes will require the development of new tools. Expected final online publication date for the, Volume 72 is May 2021. Please see http://www.annualreviews.org/page/journal/pubdates for revised estimates.
A route to de novo domestication of wild allotetraploid rice
DOI:10.1016/j.cell.2021.01.013 URL [本文引用: 1]
Earth BioGenome Project: sequencing life for the future of life
DOI:10.1073/pnas.1720115115
PMID:29686065
[本文引用: 1]
Increasing our understanding of Earth's biodiversity and responsibly stewarding its resources are among the most crucial scientific and social challenges of the new millennium. These challenges require fundamental new knowledge of the organization, evolution, functions, and interactions among millions of the planet's organisms. Herein, we present a perspective on the Earth BioGenome Project (EBP), a moonshot for biology that aims to sequence, catalog, and characterize the genomes of all of Earth's eukaryotic biodiversity over a period of 10 years. The outcomes of the EBP will inform a broad range of major issues facing humanity, such as the impact of climate change on biodiversity, the conservation of endangered species and ecosystems, and the preservation and enhancement of ecosystem services. We describe hurdles that the project faces, including data-sharing policies that ensure a permanent, freely available resource for future scientific discovery while respecting access and benefit sharing guidelines of the Nagoya Protocol. We also describe scientific and organizational challenges in executing such an ambitious project, and the structure proposed to achieve the project's goals. The far-reaching potential benefits of creating an open digital repository of genomic information for life on Earth can be realized only by a coordinated international effort.
10KP: a phylodiverse genome sequencing plan
DOI:10.1093/gigascience/giy013
PMID:29618049
[本文引用: 1]
Understanding plant evolution and diversity in a phylogenomic context is an enormous challenge due, in part, to limited availability of genome-scale data across phylodiverse species. The 10KP (10,000 Plants) Genome Sequencing Project will sequence and characterize representative genomes from every major clade of embryophytes, green algae, and protists (excluding fungi) within the next 5 years. By implementing and continuously improving leading-edge sequencing technologies and bioinformatics tools, 10KP will catalogue the genome content of plant and protist diversity and make these data freely available as an enduring foundation for future scientific discoveries and applications. 10KP is structured as an international consortium, open to the global community, including botanical gardens, plant research institutes, universities, and private industry. Our immediate goal is to establish a policy framework for this endeavor, the principles of which are outlined here.
SoyOmics: a deeply integrated database on soybean multi-omics
A multi-omics integrative network map of maize
DOI:10.1038/s41588-022-01262-1
PMID:36581701
[本文引用: 1]
Networks are powerful tools to uncover functional roles of genes in phenotypic variation at a system-wide scale. Here, we constructed a maize network map that contains the genomic, transcriptomic, translatomic and proteomic networks across maize development. This map comprises over 2.8 million edges in more than 1,400 functional subnetworks, demonstrating an extensive network divergence of duplicated genes. We applied this map to identify factors regulating flowering time and identified 2,651 genes enriched in eight subnetworks. We validated the functions of 20 genes, including 18 with previously unknown connections to flowering time in maize. Furthermore, we uncovered a flowering pathway involving histone modification. The multi-omics integrative network map illustrates the principles of how molecular networks connect different types of genes and potential pathways to map a genome-wide functional landscape in maize, which should be applicable in a wide range of species.© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Exploring structural variation and gene family architecture with De novo assemblies of 15 Medicago genomes
DOI:10.1186/s12864-017-3654-1 [本文引用: 1]
Pan-genome of cultivated pepper (Capsicum) and its use in gene presence-absence variation analyses
DOI:10.1111/nph.15413 PMID:30129229 [本文引用: 1]
Insight into the evolution and functional characteristics of the pan-genome assembly from sesame landraces and modern cultivars
DOI:10.1111/pbi.13022
PMID:30315621
[本文引用: 1]
Sesame (Sesamum indicum L.) is an important oil crop renowned for its high oil content and quality. Recently, genome assemblies for five sesame varieties including two landraces (S. indicum cv. Baizhima and Mishuozhima) and three modern cultivars (S. indicum var. Zhongzhi13, Yuzhi11 and Swetha), have become available providing a rich resource for comparative genomic analyses and gene discovery. Here, we employed a reference-assisted assembly approach to improve the draft assemblies of four of the sesame varieties. We then constructed a sesame pan-genome of 554.05 Mb. The pan-genome contained 26 472 orthologous gene clusters; 15 409 (58.21%) of them were core (present across all five sesame genomes), whereas the remaining 41.79% (11 063) clusters and the 15 890 variety-specific genes were dispensable. Comparisons between varieties suggest that modern cultivars from China and India display significant genomic variation. The gene families unique to the sesame modern cultivars contain genes mainly related to yield and quality, while those unique to the landraces contain genes involved in environmental adaptation. Comparative evolutionary analysis indicates that several genes involved in plant-pathogen interaction and lipid metabolism are under positive selection, which may be associated with sesame environmental adaption and selection for high seed oil content. This study of the sesame pan-genome provides insights into the evolution and genomic characteristics of this important oilseed and constitutes a resource for further sesame crop improvement.© 2018 The Authors. Plant Biotechnology Journal published by Society for Experimental Biology and The Association of Applied Biologists and John Wiley & Sons Ltd.
The barley pan-genome reveals the hidden legacy of mutation breeding
A chickpea genetic variation map based on the sequencing of 3,366 genomes
DOI:10.1038/s41586-021-04066-1
[本文引用: 1]
Zero hunger and good health could be realized by 2030 through effective conservation, characterization and utilization of germplasm resources1. So far, few chickpea (Cicerarietinum) germplasm accessions have been characterized at the genome sequence level2. Here we present a detailed map of variation in 3,171 cultivated and 195 wild accessions to provide publicly available resources for chickpea genomics research and breeding. We constructed a chickpea pan-genome to describe genomic diversity across cultivated chickpea and its wild progenitor accessions. A divergence tree using genes present in around 80% of individuals in one species allowed us to estimate the divergence of Cicer over the last 21 million years. Our analysis found chromosomal segments and genes that show signatures of selection during domestication, migration and improvement. The chromosomal locations of deleterious mutations responsible for limited genetic diversity and decreased fitness were identified in elite germplasm. We identified superior haplotypes for improvement-related traits in landraces that can be introgressed into elite breeding lines through haplotype-based breeding, and found targets for purging deleterious alleles through genomics-assisted breeding and/or gene editing. Finally, we propose three crop breeding strategies based on genomic prediction to enhance crop productivity for 16 traits while avoiding the erosion of genetic diversity through optimal contribution selection (OCS)-based pre-breeding. The predicted performance for 100-seed weight, an important yield-related trait, increased by up to 23% and 12% with OCS- and haplotype-based genomic approaches, respectively.
Cotton pan-genome retrieves the lost sequences and genes during domestication and selection
DOI:10.1186/s13059-021-02351-w
PMID:33892774
[本文引用: 1]
Millennia of directional human selection has reshaped the genomic architecture of cultivated cotton relative to wild counterparts, but we have limited understanding of the selective retention and fractionation of genomic components.We construct a comprehensive genomic variome based on 1961 cottons and identify 456 Mb and 357 Mb of sequence with domestication and improvement selection signals and 162 loci, 84 of which are novel, including 47 loci associated with 16 agronomic traits. Using pan-genome analyses, we identify 32,569 and 8851 non-reference genes lost from Gossypium hirsutum and Gossypium barbadense reference genomes respectively, of which 38.2% (39,278) and 14.2% (11,359) of genes exhibit presence/absence variation (PAV). We document the landscape of PAV selection accompanied by asymmetric gene gain and loss and identify 124 PAVs linked to favorable fiber quality and yield loci.This variation repertoire points to genomic divergence during cotton domestication and improvement, which informs the characterization of favorable gene alleles for improved breeding practice using a pan-genome-based approach.
Extensive variation within the pan-genome of cultivated and wild sorghum
DOI:10.1038/s41477-021-00925-x
PMID:34017083
[本文引用: 1]
Sorghum is a drought-tolerant staple crop for half a billion people in Africa and Asia, an important source of animal feed throughout the world and a biofuel feedstock of growing importance. Cultivated sorghum and its inter-fertile wild relatives constitute the primary gene pool for sorghum. Understanding and characterizing the diversity within this valuable resource is fundamental for its effective utilization in crop improvement. Here, we report analysis of a sorghum pan-genome to explore genetic diversity within the sorghum primary gene pool. We assembled 13 genomes representing cultivated sorghum and its wild relatives, and integrated them with 3 other published genomes to generate a pan-genome of 44,079 gene families with 222.6 Mb of new sequence identified. The pan-genome displays substantial gene-content variation, with 64% of gene families showing presence/absence variation among genomes. Comparisons between core genes and dispensable genes suggest that dispensable genes are important for sorghum adaptation. Extensive genetic variation was uncovered within the pan-genome, and the distribution of these variations was influenced by variation of recombination rate and transposable element content across the genome. We identified presence/absence variants that were under selection during sorghum domestication and improvement, and demonstrated that such variation had important phenotypic outcomes that could contribute to crop improvement. The constructed sorghum pan-genome represents an important resource for sorghum improvement and gene discovery.
De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes
Pan-genome of Raphanus highlights genetic variation and introgression among domesticated, wild, and weedy radishes
DOI:10.1016/j.molp.2021.08.005 URL [本文引用: 1]
Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species
DOI:10.1038/s41588-023-01340-y
PMID:37024581
[本文引用: 2]
Effective utilization of wild relatives is key to overcoming challenges in genetic improvement of cultivated tomato, which has a narrow genetic basis; however, current efforts to decipher high-quality genomes for tomato wild species are insufficient. Here, we report chromosome-scale tomato genomes from nine wild species and two cultivated accessions, representative of Solanum section Lycopersicon, the tomato clade. Together with two previously released genomes, we elucidate the phylogeny of Lycopersicon and construct a section-wide gene repertoire. We reveal the landscape of structural variants and provide entry to the genomic diversity among tomato wild relatives, enabling the discovery of a wild tomato gene with the potential to increase yields of modern cultivated tomatoes. Construction of a graph-based genome enables structural-variant-based genome-wide association studies, identifying numerous signals associated with tomato flavor-related traits and fruit metabolites. The tomato super-pangenome resources will expedite biological studies and breeding of this globally important crop.© 2023. The Author(s).
Genomic innovation and regulatory rewiring during evolution of the cotton genus Gossypium
DOI:10.1038/s41588-022-01237-2
PMID:36474047
[本文引用: 1]
Phenotypic diversity and evolutionary innovation ultimately trace to variation in genomic sequence and rewiring of regulatory networks. Here, we constructed a pan-genome of the Gossypium genus using ten representative diploid genomes. We document the genomic evolutionary history and the impact of lineage-specific transposon amplification on differential genome composition. The pan-3D genome reveals evolutionary connections between transposon-driven genome size variation and both higher-order chromatin structure reorganization and the rewiring of chromatin interactome. We linked changes in chromatin structures to phenotypic differences in cotton fiber and identified regulatory variations that decode the genetic basis of fiber length, the latter enabled by sequencing 1,005 transcriptomes during fiber development. We showcase how pan-genomic, pan-3D genomic and genetic regulatory data serve as a resource for delineating the evolutionary basis of spinnable cotton fiber. Our work provides insights into the evolution of genome organization and regulation and will inform cotton improvement by enabling regulome-based approaches.© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Genome evolution and diversity of wild and cultivated potatoes
DOI:10.1038/s41586-022-04822-x
[本文引用: 1]
Potato (Solanum tuberosum L.) is the world’s most important non-cereal food crop, and the vast majority of commercially grown cultivars are highly heterozygous tetraploids. Advances in diploid hybrid breeding based on true seeds have the potential to revolutionize future potato breeding and production1–4. So far, relatively few studies have examined the genome evolution and diversity of wild and cultivated landrace potatoes, which limits the application of their diversity in potato breeding. Here we assemble 44 high-quality diploid potato genomes from 24 wild and 20 cultivated accessions that are representative of Solanum section Petota, the tuber-bearing clade, as well as 2 genomes from the neighbouring section, Etuberosum. Extensive discordance of phylogenomic relationships suggests the complexity of potato evolution. We find that the potato genome substantially expanded its repertoire of disease-resistance genes when compared with closely related seed-propagated solanaceous crops, indicative of the effect of tuber-based propagation strategies on the evolution of the potato genome. We discover a transcription factor that determines tuber identity and interacts with the mobile tuberization inductive signal SP6A. We also identify 561,433 high-confidence structural variants and construct a map of large inversions, which provides insights for improving inbred lines and precluding potential linkage drag, as exemplified by a 5.8-Mb inversion that is associated with carotenoid content in tubers. This study will accelerate hybrid potato breeding and enrich our understanding of the evolution and biology of potato as a global staple food crop.
De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis
DOI:10.1038/s41588-022-01283-w [本文引用: 1]



{kind=link}
{kind=link}