Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors
1
2010
... 无义介导的mRNA降解(nonsense-mediated mRNA decay, NMD)是一种广泛存在于真核生物的mRNA质量监控机制,特异性识别并降解含有提前终止密码子(premature termination codon, PTC)的mRNA[1],同时NMD也是机体调控基因表达的重要方式之一.在正常生理情况下,mRNA的可变剪接、转录错误或程序性基因重排可产生PTC,触发NMD途径使其降解,从而阻止有害的截短蛋白的产生.在病理情况下,以无义和移码变异为主的基因突变也可产生PTC,通过触发NMD降解mRNA导致靶蛋白表达下降从而引发疾病[2,3].约1/3已知病因的人类遗传病是由基因突变产生的PTC所致[4]. ...
Regulation of cytoplasmic mRNA decay
1
2012
... 无义介导的mRNA降解(nonsense-mediated mRNA decay, NMD)是一种广泛存在于真核生物的mRNA质量监控机制,特异性识别并降解含有提前终止密码子(premature termination codon, PTC)的mRNA[1],同时NMD也是机体调控基因表达的重要方式之一.在正常生理情况下,mRNA的可变剪接、转录错误或程序性基因重排可产生PTC,触发NMD途径使其降解,从而阻止有害的截短蛋白的产生.在病理情况下,以无义和移码变异为主的基因突变也可产生PTC,通过触发NMD降解mRNA导致靶蛋白表达下降从而引发疾病[2,3].约1/3已知病因的人类遗传病是由基因突变产生的PTC所致[4]. ...
NMD: At the crossroads between translation termination and ribosome recycling
3
2015
... 无义介导的mRNA降解(nonsense-mediated mRNA decay, NMD)是一种广泛存在于真核生物的mRNA质量监控机制,特异性识别并降解含有提前终止密码子(premature termination codon, PTC)的mRNA[1],同时NMD也是机体调控基因表达的重要方式之一.在正常生理情况下,mRNA的可变剪接、转录错误或程序性基因重排可产生PTC,触发NMD途径使其降解,从而阻止有害的截短蛋白的产生.在病理情况下,以无义和移码变异为主的基因突变也可产生PTC,通过触发NMD降解mRNA导致靶蛋白表达下降从而引发疾病[2,3].约1/3已知病因的人类遗传病是由基因突变产生的PTC所致[4]. ...
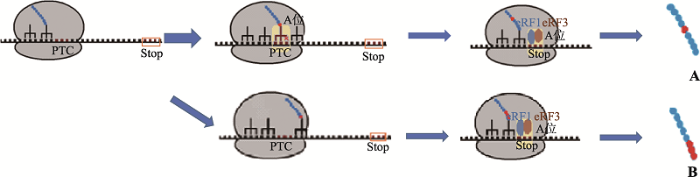
... 因此,蛋白质的翻译是终止还是继续取决于释放因子和非同工tRNA之间相互竞争与核糖体A位点的结合[3].当释放因子识别PTC并与核糖体大亚基的A位结合时,则翻译终止,触发NMD途径,mRNA发生降解.虽然细胞内存在多种校正机制以确保mRNA上的密码子可以被正确识别,但是准确率仍然达不到100%.当携带某种氨基酸的非同工tRNA与核糖体A位偶然结合,使无义密码子重新被解码为有义密码子,PTC被该氨基酸所取代,翻译继续进行,即表现为由于tRNA错配导致的PTC通读(图1A).此外,通常情况下核糖体对mRNA密码子的识别是由起始密码子开始,沿5′至3′端方向,一个接一个直到终止密码子,但也有一些例外,翻译过程中某些反式作用因子决定核糖体在mRNA特定位点上的读码框可以发生程序性移动一个或者多个核苷酸后继续翻译,这一过程称为程序性核糖体移码或核糖体跳跃(ribosome frameshifting/jumping) (图1B)[12].当核糖体恰巧跳过了PTC,则引发由于核糖体跳跃导致的PTC通读,但是目前尚缺少相关报道. ...
... 蛋白质翻译终止后,留下一个由mRNA、位于P位的去酰化-tRNA和核糖体组成的翻译终止后核糖体复合物.随后,在ATP酶ABCE1、起始因子eIF3、eIF1、eIF1A以及eIF3j的协同作用下,翻译终止后核糖体复合物解体为mRNA、去酰化-tRNA和核糖体单体或其亚基[3].解体后的核糖体进入新一轮的蛋白质合成. ...
NMD: RNA biology meets human genetic medicine
1
2010
... 无义介导的mRNA降解(nonsense-mediated mRNA decay, NMD)是一种广泛存在于真核生物的mRNA质量监控机制,特异性识别并降解含有提前终止密码子(premature termination codon, PTC)的mRNA[1],同时NMD也是机体调控基因表达的重要方式之一.在正常生理情况下,mRNA的可变剪接、转录错误或程序性基因重排可产生PTC,触发NMD途径使其降解,从而阻止有害的截短蛋白的产生.在病理情况下,以无义和移码变异为主的基因突变也可产生PTC,通过触发NMD降解mRNA导致靶蛋白表达下降从而引发疾病[2,3].约1/3已知病因的人类遗传病是由基因突变产生的PTC所致[4]. ...
无义介导的mRNA降解机制及其在单基因遗传病中的作用
1
2012
... 在哺乳动物细胞中,前体mRNA被剪接后在距离外显子与外显子连接点(exon-exon junction, EEJ)上游约20~24个核苷酸的位置处会形成一个外显子-外显子连接点复合物(exon-exon junction complex, EJC).在正常的翻译过程中,核糖体沿着mRNA移动,替换下游所有的EJC并在正常的终止密码子处停止翻译,产生全长蛋白.若mRNA中存在PTC,翻译核糖体会在PTC处停止,募集UPF1、SMG1和两个翻译释放因子(eRF1和eRF3)形成SURF复合物.该复合物通过对下游EJC的识别,使NMD的核心组分-UPF1、UPF2和UPF3相互作用,最终导致UPF1发生磷酸化.磷酸化的UPF1一方面靶向作用于翻译起始因子eIF3抑制下一轮翻译的开始,另一方面进一步招募具有核酸内切酶和核酸外切酶活性的SMG系列因子介导mRNA的降解[5].NMD效率在不同的转录本、细胞和组织类型以及个体间均存在广泛的变异性,更有甚者甚至不发生NDM,即产生了所谓的NMD逃逸(NMD escape)现象.本文对NMD逃逸的机制及其在临床中的应用进行了综述. ...
无义介导的mRNA降解机制及其在单基因遗传病中的作用
1
2012
... 在哺乳动物细胞中,前体mRNA被剪接后在距离外显子与外显子连接点(exon-exon junction, EEJ)上游约20~24个核苷酸的位置处会形成一个外显子-外显子连接点复合物(exon-exon junction complex, EJC).在正常的翻译过程中,核糖体沿着mRNA移动,替换下游所有的EJC并在正常的终止密码子处停止翻译,产生全长蛋白.若mRNA中存在PTC,翻译核糖体会在PTC处停止,募集UPF1、SMG1和两个翻译释放因子(eRF1和eRF3)形成SURF复合物.该复合物通过对下游EJC的识别,使NMD的核心组分-UPF1、UPF2和UPF3相互作用,最终导致UPF1发生磷酸化.磷酸化的UPF1一方面靶向作用于翻译起始因子eIF3抑制下一轮翻译的开始,另一方面进一步招募具有核酸内切酶和核酸外切酶活性的SMG系列因子介导mRNA的降解[5].NMD效率在不同的转录本、细胞和组织类型以及个体间均存在广泛的变异性,更有甚者甚至不发生NDM,即产生了所谓的NMD逃逸(NMD escape)现象.本文对NMD逃逸的机制及其在临床中的应用进行了综述. ...
A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance
1
1998
... 通常,当PTC距离最后一个EEJ大于或等于50~55个核苷酸即可触发NMD,称为NMD途径的50边界规则[6].然而,研究者发现某些PTC虽然符合这个规则,但是却没有发生NMD导致mRNA的降解,学者称这种现象为NMD逃逸.通过分析功能基因组学数据库发现有52.7%的人群常见的无义变异发生了NMD逃逸[7].研究表明这些发生NMD逃逸的转录本,其PTC多位于距离翻译起始位点较近的区域,比如一些位于人β球蛋白基因外显子1的PTC并未触发NMD而导致mRNA的降解,携带这类PTC的转录本与野生型β球蛋白mRNA转录水平相当[8].这种现象也被称为“AUG邻近效应”[9,10]:即在加帽介导的翻译起始过程中,胞质多聚腺苷酸结合蛋白1 (PABPC1)通过与真核起始因子eIF4G和eIF3之间的相互作用,靠近40S核糖体亚基,形成环状结构.PABPC1也因此邻近AUG起始密码子.随着翻译延伸的开始,这个闭合的环状结构使PABPC1与位于AUG附近PTC上的终止复合物近在咫尺.此时PABPC1竞争性的与终止复合物中的eRF3相互作用,从而削弱了eRF3与NMD关键因子UPF1间的作用,抑制NMD途径. ...
Translational plasticity facilitates the accumulation of nonsense genetic variants in the human population
2
2016
... 通常,当PTC距离最后一个EEJ大于或等于50~55个核苷酸即可触发NMD,称为NMD途径的50边界规则[6].然而,研究者发现某些PTC虽然符合这个规则,但是却没有发生NMD导致mRNA的降解,学者称这种现象为NMD逃逸.通过分析功能基因组学数据库发现有52.7%的人群常见的无义变异发生了NMD逃逸[7].研究表明这些发生NMD逃逸的转录本,其PTC多位于距离翻译起始位点较近的区域,比如一些位于人β球蛋白基因外显子1的PTC并未触发NMD而导致mRNA的降解,携带这类PTC的转录本与野生型β球蛋白mRNA转录水平相当[8].这种现象也被称为“AUG邻近效应”[9,10]:即在加帽介导的翻译起始过程中,胞质多聚腺苷酸结合蛋白1 (PABPC1)通过与真核起始因子eIF4G和eIF3之间的相互作用,靠近40S核糖体亚基,形成环状结构.PABPC1也因此邻近AUG起始密码子.随着翻译延伸的开始,这个闭合的环状结构使PABPC1与位于AUG附近PTC上的终止复合物近在咫尺.此时PABPC1竞争性的与终止复合物中的eRF3相互作用,从而削弱了eRF3与NMD关键因子UPF1间的作用,抑制NMD途径. ...
... Jagannathan等[7]利用生物信息学方法分析了人群中单核苷酸变异下游50个密码子的序列,研究发现与同义变异相比,Met在无义变异下游的50个密码子内存在显著的富集:57%的罕见和85%的常见无义变异在其下游50个密码子内存在Met,即潜在的翻译起始点;而只有52%的同义变异存在该现象,且这一富集现象仅在编码序列的前10%中成立.以上研究提示,有效的翻译重新启动与正常的起始密码子、PTC和潜在起始密码子3者之间的距离有关.首先,翻译终止后的核糖体与正常的起始密码子足够接近,也就是PTC需要靠近正常的翻译起始密码子;其次PTC距离下游的潜在起始点也较近,使得核糖体可以相对较快的遇到起始位点重启翻译. ...
Mechanism of escape from nonsense-mediated mRNA decay of human β-globin transcripts with nonsense mutations in the first exon
1
2011
... 通常,当PTC距离最后一个EEJ大于或等于50~55个核苷酸即可触发NMD,称为NMD途径的50边界规则[6].然而,研究者发现某些PTC虽然符合这个规则,但是却没有发生NMD导致mRNA的降解,学者称这种现象为NMD逃逸.通过分析功能基因组学数据库发现有52.7%的人群常见的无义变异发生了NMD逃逸[7].研究表明这些发生NMD逃逸的转录本,其PTC多位于距离翻译起始位点较近的区域,比如一些位于人β球蛋白基因外显子1的PTC并未触发NMD而导致mRNA的降解,携带这类PTC的转录本与野生型β球蛋白mRNA转录水平相当[8].这种现象也被称为“AUG邻近效应”[9,10]:即在加帽介导的翻译起始过程中,胞质多聚腺苷酸结合蛋白1 (PABPC1)通过与真核起始因子eIF4G和eIF3之间的相互作用,靠近40S核糖体亚基,形成环状结构.PABPC1也因此邻近AUG起始密码子.随着翻译延伸的开始,这个闭合的环状结构使PABPC1与位于AUG附近PTC上的终止复合物近在咫尺.此时PABPC1竞争性的与终止复合物中的eRF3相互作用,从而削弱了eRF3与NMD关键因子UPF1间的作用,抑制NMD途径. ...
Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG-proximal nonsense mutations
1
2012
... 通常,当PTC距离最后一个EEJ大于或等于50~55个核苷酸即可触发NMD,称为NMD途径的50边界规则[6].然而,研究者发现某些PTC虽然符合这个规则,但是却没有发生NMD导致mRNA的降解,学者称这种现象为NMD逃逸.通过分析功能基因组学数据库发现有52.7%的人群常见的无义变异发生了NMD逃逸[7].研究表明这些发生NMD逃逸的转录本,其PTC多位于距离翻译起始位点较近的区域,比如一些位于人β球蛋白基因外显子1的PTC并未触发NMD而导致mRNA的降解,携带这类PTC的转录本与野生型β球蛋白mRNA转录水平相当[8].这种现象也被称为“AUG邻近效应”[9,10]:即在加帽介导的翻译起始过程中,胞质多聚腺苷酸结合蛋白1 (PABPC1)通过与真核起始因子eIF4G和eIF3之间的相互作用,靠近40S核糖体亚基,形成环状结构.PABPC1也因此邻近AUG起始密码子.随着翻译延伸的开始,这个闭合的环状结构使PABPC1与位于AUG附近PTC上的终止复合物近在咫尺.此时PABPC1竞争性的与终止复合物中的eRF3相互作用,从而削弱了eRF3与NMD关键因子UPF1间的作用,抑制NMD途径. ...
Resistance of mRNAs with AUG-proximal nonsense mutations to nonsense-mediated decay reflects variables of mRNA structure and translational activity
2
2015
... 通常,当PTC距离最后一个EEJ大于或等于50~55个核苷酸即可触发NMD,称为NMD途径的50边界规则[6].然而,研究者发现某些PTC虽然符合这个规则,但是却没有发生NMD导致mRNA的降解,学者称这种现象为NMD逃逸.通过分析功能基因组学数据库发现有52.7%的人群常见的无义变异发生了NMD逃逸[7].研究表明这些发生NMD逃逸的转录本,其PTC多位于距离翻译起始位点较近的区域,比如一些位于人β球蛋白基因外显子1的PTC并未触发NMD而导致mRNA的降解,携带这类PTC的转录本与野生型β球蛋白mRNA转录水平相当[8].这种现象也被称为“AUG邻近效应”[9,10]:即在加帽介导的翻译起始过程中,胞质多聚腺苷酸结合蛋白1 (PABPC1)通过与真核起始因子eIF4G和eIF3之间的相互作用,靠近40S核糖体亚基,形成环状结构.PABPC1也因此邻近AUG起始密码子.随着翻译延伸的开始,这个闭合的环状结构使PABPC1与位于AUG附近PTC上的终止复合物近在咫尺.此时PABPC1竞争性的与终止复合物中的eRF3相互作用,从而削弱了eRF3与NMD关键因子UPF1间的作用,抑制NMD途径. ...
... 已有报道邻近翻译起始密码子的PTC,由于在其下游重新启动了有效的翻译导致携带该PTC的mRNA转录本发生了NMD逃逸,包括神经鞘瘤相关基因SMARCB1[17]、乳腺癌相关基因BRCA1、LQT2基因[18]、α和β珠蛋白基因[10]等,提示有效的翻译重新启动可能存在着边界效应,位于边界内的PTC可通过翻译重新启动而逃离NMD,而边界外的PTC则不能;此外,不同基因转录本的重新启动,其边界也有所不同,即便是密切相关的α和β珠蛋白基因也是如此. ...
Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases
1
2011
... PTC通读是指在蛋白翻译过程中,由于tRNA错配或核糖体移码或跳跃等原因导致翻译可以顺利通过PTC,直至遇到正常的终止密码子.就通读效率而言,PTC的通读效率约为正常终止密码子的10倍,因而哺乳动物细胞中通读更倾向于发生在PTC,而对于正常终止密码子的通读则极为罕见且低效[11]. ...
Mechanisms of mRNA frame maintenance and its subversion during translation of the genetic code
1
2015
... 因此,蛋白质的翻译是终止还是继续取决于释放因子和非同工tRNA之间相互竞争与核糖体A位点的结合[3].当释放因子识别PTC并与核糖体大亚基的A位结合时,则翻译终止,触发NMD途径,mRNA发生降解.虽然细胞内存在多种校正机制以确保mRNA上的密码子可以被正确识别,但是准确率仍然达不到100%.当携带某种氨基酸的非同工tRNA与核糖体A位偶然结合,使无义密码子重新被解码为有义密码子,PTC被该氨基酸所取代,翻译继续进行,即表现为由于tRNA错配导致的PTC通读(图1A).此外,通常情况下核糖体对mRNA密码子的识别是由起始密码子开始,沿5′至3′端方向,一个接一个直到终止密码子,但也有一些例外,翻译过程中某些反式作用因子决定核糖体在mRNA特定位点上的读码框可以发生程序性移动一个或者多个核苷酸后继续翻译,这一过程称为程序性核糖体移码或核糖体跳跃(ribosome frameshifting/jumping) (图1B)[12].当核糖体恰巧跳过了PTC,则引发由于核糖体跳跃导致的PTC通读,但是目前尚缺少相关报道. ...
Sense from nonsense: therapies for premature stop codon diseases
1
2012
... 综上所述,因PTC通读引发的NMD逃逸,具有以下两个特点:首先,由于未触发NMD途径,因此携带PTC转录本的转录水平与野生型相当;其次,由于PTC被氨基酸取代,且翻译终止于正常的终止密码子,因而可产生全长的蛋白质.虽然取代PTC的氨基酸可能与正常蛋白相应位置的氨基酸不同,但是如果该位置不是蛋白的关键功能区域,那么新生成的蛋白可发挥正常或部分正常功能.因此,这一机制也是目前主要用于抑制病理性NMD的有害作用、上调功能蛋白的表达从而靶向治疗遗传性疾病的分子基础[13],使用氨基糖甙类抗生素、非氨基糖甙类抗生素、小分子化合物(如PTC124)以及可与PTC结合的tRNA衍生物等均可诱导PTC的通读[14]. ...
促通读药物的作用机制与临床应用
1
2016
... 综上所述,因PTC通读引发的NMD逃逸,具有以下两个特点:首先,由于未触发NMD途径,因此携带PTC转录本的转录水平与野生型相当;其次,由于PTC被氨基酸取代,且翻译终止于正常的终止密码子,因而可产生全长的蛋白质.虽然取代PTC的氨基酸可能与正常蛋白相应位置的氨基酸不同,但是如果该位置不是蛋白的关键功能区域,那么新生成的蛋白可发挥正常或部分正常功能.因此,这一机制也是目前主要用于抑制病理性NMD的有害作用、上调功能蛋白的表达从而靶向治疗遗传性疾病的分子基础[13],使用氨基糖甙类抗生素、非氨基糖甙类抗生素、小分子化合物(如PTC124)以及可与PTC结合的tRNA衍生物等均可诱导PTC的通读[14]. ...
促通读药物的作用机制与临床应用
1
2016
... 综上所述,因PTC通读引发的NMD逃逸,具有以下两个特点:首先,由于未触发NMD途径,因此携带PTC转录本的转录水平与野生型相当;其次,由于PTC被氨基酸取代,且翻译终止于正常的终止密码子,因而可产生全长的蛋白质.虽然取代PTC的氨基酸可能与正常蛋白相应位置的氨基酸不同,但是如果该位置不是蛋白的关键功能区域,那么新生成的蛋白可发挥正常或部分正常功能.因此,这一机制也是目前主要用于抑制病理性NMD的有害作用、上调功能蛋白的表达从而靶向治疗遗传性疾病的分子基础[13],使用氨基糖甙类抗生素、非氨基糖甙类抗生素、小分子化合物(如PTC124)以及可与PTC结合的tRNA衍生物等均可诱导PTC的通读[14]. ...
Translational read-through of a nonsense mutation in ATP7A impacts treatment outcome in Menkes disease
2
2009
... Menkes综合征是一种由ATP7A基因突变导致铜转运障碍而引起的多系统受累的X连锁隐性遗传病,患者主要表现为神经系统变性和结缔组织异常.ATP7A基因位于Xq13.3,包含23个外显子,编码1500个氨基酸的蛋白产物ATP7A.Kaler等[15]报道了1例Menkes综合征患者携带有c.746C>T (p.Arg201Ter)无义变异,该变异在ATP7A基因的第3外显子产生一个PTC.患者cDNA的Sanger测序在相应位置可见明显C>T的碱基替换峰,提示携带PTC的mRNA转录本未发生显著的降解;应用Western blot和免疫组化分析患儿细胞,均可检测到少量全长ATP7A蛋白.以上的检测结果符合由于通读机制导致的NMD逃逸特点.因此,作者推断携带c.746C>T (p.Arg201Ter)无义变异的转录本发生了通读导致的NMD逃逸,为了进一步明确该通读是否源于tRNA错配,作者试图检测氨基酸序列,遗憾的是此例患者NMD抑制效率不是很高,未能足量分离和纯化通读产物.但是,酵母互补实验揭示该突变体表达的蛋白仍保留有部分铜转运功能,提示PTC的通读产物保留了部分功能.加之该患者在出生后不久即被诊断并进行铜注射治疗,因此治疗效果显著,无明显的神经系统症状出现[15]. ...
... [15]. ...
Please do not recycle! Translation reinitiation in microbes and higher eukaryotes
1
2018
... 翻译的重新启动是指在PTC的下游存在着AUG,即潜在的翻译起始点.由于Met的三联密码同为AUG与起始密码子相同,因此可作为PTC下游翻译重新启动的潜在起始点.翻译终止后,核糖体复合物并未完全解离进入再循环,而是沿着mRNA继续滑动,当它遇到潜在的翻译起始点时,蛋白质的翻译重新开始[16]. ...
Premature termination of SMARCB1 translation may be followed by reinitiation in schwannomatosis- associated schwannomas, but results in absence of SMARCB1 expression in rhabdoid tumors
1
2014
... 已有报道邻近翻译起始密码子的PTC,由于在其下游重新启动了有效的翻译导致携带该PTC的mRNA转录本发生了NMD逃逸,包括神经鞘瘤相关基因SMARCB1[17]、乳腺癌相关基因BRCA1、LQT2基因[18]、α和β珠蛋白基因[10]等,提示有效的翻译重新启动可能存在着边界效应,位于边界内的PTC可通过翻译重新启动而逃离NMD,而边界外的PTC则不能;此外,不同基因转录本的重新启动,其边界也有所不同,即便是密切相关的α和β珠蛋白基因也是如此. ...
LQT2 nonsense mutations generate trafficking defective NH2-terminally truncated channels by the reinitiation of translation
1
2013
... 已有报道邻近翻译起始密码子的PTC,由于在其下游重新启动了有效的翻译导致携带该PTC的mRNA转录本发生了NMD逃逸,包括神经鞘瘤相关基因SMARCB1[17]、乳腺癌相关基因BRCA1、LQT2基因[18]、α和β珠蛋白基因[10]等,提示有效的翻译重新启动可能存在着边界效应,位于边界内的PTC可通过翻译重新启动而逃离NMD,而边界外的PTC则不能;此外,不同基因转录本的重新启动,其边界也有所不同,即便是密切相关的α和β珠蛋白基因也是如此. ...
Does eIF3 promote reinitiation after translation of short upstream ORFs also in mammalian cells?
1
2017
... 尽管翻译的重新启动一直被认为是NMD逃逸的重要原因,但其发生的具体机制尚不十分清楚.具有短uORF转录本的翻译后重新启动是机体应对环境变化调节基因表达的途径之一.研究发现这类转录本在翻译终止后,只有核糖体的大亚基和去酰化tRNA被循环再利用,小亚基仍保留在mRNA上,进入下游从而发生翻译的重新启动.进一步研究显示真核生物起始因子eIF3,在翻译的延伸和终止过程中可与核糖体发生短暂结合[19,20],eIF3可特异性稳定小亚基,使其不被循环,保留eIF3的核糖体能够继续向下游移动,识别PTC下游的潜在起始密码子重新启动翻译(图2).综上所述,因翻译的重新启动引发的NMD逃逸,同样由于未触发NMD途径,携带PTC转录本的转录水平也与野生型相当;其次,翻译重新启动发生于PTC下游的潜在起始点,因而可产生N端截短蛋白. ...
In vivo evidence that eIF3 stays bound to ribosomes elongating and terminating on short upstream ORFs to promote reinitiation
1
2017
... 尽管翻译的重新启动一直被认为是NMD逃逸的重要原因,但其发生的具体机制尚不十分清楚.具有短uORF转录本的翻译后重新启动是机体应对环境变化调节基因表达的途径之一.研究发现这类转录本在翻译终止后,只有核糖体的大亚基和去酰化tRNA被循环再利用,小亚基仍保留在mRNA上,进入下游从而发生翻译的重新启动.进一步研究显示真核生物起始因子eIF3,在翻译的延伸和终止过程中可与核糖体发生短暂结合[19,20],eIF3可特异性稳定小亚基,使其不被循环,保留eIF3的核糖体能够继续向下游移动,识别PTC下游的潜在起始密码子重新启动翻译(图2).综上所述,因翻译的重新启动引发的NMD逃逸,同样由于未触发NMD途径,携带PTC转录本的转录水平也与野生型相当;其次,翻译重新启动发生于PTC下游的潜在起始点,因而可产生N端截短蛋白. ...
BRCA1 in breast and ovarian cancer predisposition
1
2005
... BRCA1基因与乳腺癌的发生高度相关,该基因发生突变使女性容易罹患乳腺癌和卵巢癌.该基因定位于17q21,含有23个外显子,在DNA修复、细胞周期调控、蛋白质泛素化和染色质重塑中发挥重要作用[21].Buisson等[22]研究发现BRCA1基因的移码变异185delAG (c.68_69delAG)和188del11 (c.71_81del),分别在第36位(Ter36)和39(Ter39)位氨基酸位置产生PTC,且均位于第3外显子理论上应该触发NMD,但是Northern blot检测显示携带突变的转录本与野生型表达量相当;Western blot检测发现Ter36与Ter39突变质粒可以产生N端截短蛋白.以上检测结果符合由翻译的重新启动所致的NMD逃逸.进一步分析BRCA1基因序列显示在PTC下游22(Met48)和262(Met128)核苷酸的位置分别存在有Met,即潜在的翻译起始位点.为了明确翻译重新启动的具体位置,研究者利用定点诱变技术分别将Ter36与Ter39突变体的Met48和Met128突变为缬氨酸转染细胞,Western blot发现当Met48突变为缬氨酸时,Ter36+Met48Val与Ter39+Met48Val突变体产生的蛋白条带与均与Met48突变之前一致;而当Met128突变为缬氨酸时,Ter36+Met128Val与Ter39+Met128Val突变体则不产生N端截短蛋白.由此推测185delAG(c.68_69delAG)和188del11 (c.71_81del)变异发生NMD逃逸是原于在PTC下游的Met128位置发生了翻译的重新启动. ...
The 185delAG mutation (c.68_69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon
1
2006
... BRCA1基因与乳腺癌的发生高度相关,该基因发生突变使女性容易罹患乳腺癌和卵巢癌.该基因定位于17q21,含有23个外显子,在DNA修复、细胞周期调控、蛋白质泛素化和染色质重塑中发挥重要作用[21].Buisson等[22]研究发现BRCA1基因的移码变异185delAG (c.68_69delAG)和188del11 (c.71_81del),分别在第36位(Ter36)和39(Ter39)位氨基酸位置产生PTC,且均位于第3外显子理论上应该触发NMD,但是Northern blot检测显示携带突变的转录本与野生型表达量相当;Western blot检测发现Ter36与Ter39突变质粒可以产生N端截短蛋白.以上检测结果符合由翻译的重新启动所致的NMD逃逸.进一步分析BRCA1基因序列显示在PTC下游22(Met48)和262(Met128)核苷酸的位置分别存在有Met,即潜在的翻译起始位点.为了明确翻译重新启动的具体位置,研究者利用定点诱变技术分别将Ter36与Ter39突变体的Met48和Met128突变为缬氨酸转染细胞,Western blot发现当Met48突变为缬氨酸时,Ter36+Met48Val与Ter39+Met48Val突变体产生的蛋白条带与均与Met48突变之前一致;而当Met128突变为缬氨酸时,Ter36+Met128Val与Ter39+Met128Val突变体则不产生N端截短蛋白.由此推测185delAG(c.68_69delAG)和188del11 (c.71_81del)变异发生NMD逃逸是原于在PTC下游的Met128位置发生了翻译的重新启动. ...
A novel de novo CDH1 germline variant aids in the classification of carboxy-terminal E-cadherin alterations predicted to escape nonsense- mediated mRNA decay
1
2018
... 除PTC通读和翻译的重新启动外,还存在其他一些因素可能导致PTC的NMD逃逸[23,24],如mRNA的剪接.当mRNA的选择性剪接将包含PTC的外显子从转录本中去除,则不再发生NMD.同样,如果PTC下游的某个单一内含子拼接效率低下,PTC下游没有EJC来发出NMD信号,从而无法触发NMD.其他影响NMD效率的因素还有3′非翻译区(UTR)的序列和结构.一些含有长3′ UTR的mRNA中存在着抑制NMD的顺式作用元件.研究发现位于TRAM1基因mRNA的3′UTR前200个核苷酸的位置存在一个顺式作用元件,当其位于终止密码子下游附近时可以抑制NMD的发生[25].此外,由于NMD是一个翻译依赖的过程,影响翻译的顺式作用元件和反式作用因子均可以影响NMD的结局. ...
Stop codon readthrough generates a C-terminally extended variant of the human vitamin D receptor with reduced calcitriol response
1
2018
... 除PTC通读和翻译的重新启动外,还存在其他一些因素可能导致PTC的NMD逃逸[23,24],如mRNA的剪接.当mRNA的选择性剪接将包含PTC的外显子从转录本中去除,则不再发生NMD.同样,如果PTC下游的某个单一内含子拼接效率低下,PTC下游没有EJC来发出NMD信号,从而无法触发NMD.其他影响NMD效率的因素还有3′非翻译区(UTR)的序列和结构.一些含有长3′ UTR的mRNA中存在着抑制NMD的顺式作用元件.研究发现位于TRAM1基因mRNA的3′UTR前200个核苷酸的位置存在一个顺式作用元件,当其位于终止密码子下游附近时可以抑制NMD的发生[25].此外,由于NMD是一个翻译依赖的过程,影响翻译的顺式作用元件和反式作用因子均可以影响NMD的结局. ...
Identification of elements in human long 3' UTRs that inhibit nonsense-mediated decay
1
2015
... 除PTC通读和翻译的重新启动外,还存在其他一些因素可能导致PTC的NMD逃逸[23,24],如mRNA的剪接.当mRNA的选择性剪接将包含PTC的外显子从转录本中去除,则不再发生NMD.同样,如果PTC下游的某个单一内含子拼接效率低下,PTC下游没有EJC来发出NMD信号,从而无法触发NMD.其他影响NMD效率的因素还有3′非翻译区(UTR)的序列和结构.一些含有长3′ UTR的mRNA中存在着抑制NMD的顺式作用元件.研究发现位于TRAM1基因mRNA的3′UTR前200个核苷酸的位置存在一个顺式作用元件,当其位于终止密码子下游附近时可以抑制NMD的发生[25].此外,由于NMD是一个翻译依赖的过程,影响翻译的顺式作用元件和反式作用因子均可以影响NMD的结局. ...
Suppression of premature termination codons as a therapeutic approach
1
2012
... PTC通读的治疗首个突破发生在20世纪晚期,研究者发现一些化合物能够在序列中引入一种天然氨基酸取代PTC,从而发生通读产生完整的多肽链,避免因触发NMD导致的mRNA降解[26].随着研究的深入,越来越多的药物被确定使用(表1).目前比较成功的治疗方法主要有两类,即药物和分子生物学干预,用于治疗PTC引起的疾病. ...
Effect of readthrough treatment in fibroblasts of patients affected by lysosomal diseases caused by premature termination codons
1
2015
... Current research status of drugs
Table 1 | 通读药物 | 实验模型 | 临床试验 | 相关疾病 | 特点与问题 |
| 氨基糖苷类 | | | | |
| 庆大霉素[27] | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、法布里病(Fabry)、尼曼匹克症A/B型(NPA/NPB)、全身性神经节苷脂贮积(Gangliosidosis I )、粘多糖累积症II型(MPS II) 、MPS I-H,MPS IIIB,MPS VI、血友病A和B(HA和HB)、无脉络膜症(Choroideremia)、先天性黑朦(LCA type 2)、眼缺损(Ocular coloboma )和Usher综合征I型(USH1) | 副作用如耳毒性、肾毒性和视网膜毒性 |
| 阿米卡星[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、粘多糖
累积症I-H(MPS I-H)和脊髓型肌萎缩症I型
(SMA I) | |
| 妥布霉素[29] | 动物 | 无 | 囊性纤维化和脊髓型肌萎缩症I型(SMA I) | |
| 巴龙霉素[30] | 细胞 | 无 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、
瑞特综合征(RTT)、无脉络膜症(Choroideremia)、粘多糖累积症I-H(MPS I-H)、眼缺损(Ocular
coloboma )和Usher综合征I型(USH1) | |
| G418[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、粘多糖累积症I-H(MPS I-H)、脊髓型肌萎缩症I型(SMA I)、先天性黑朦(LCA type 2)、色素性视网膜炎(RP)和Usher综合征I型(USH1) | |
| 氨基糖苷类衍生物 | | | | |
新霉素衍生物
(TC003和TC007)[29] | 动物和细胞 | 无 | 脊髓型肌萎缩症I型(SMA I) | 生物相容性更好 |
卡那霉素衍生物
(JL023)[29] | | | 脊髓型肌萎缩症I型(SMA I) | 无明显副作用 |
巴龙霉素衍生物
(NB30、NB54、
NB84和NB124)[31] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT) Usher综合征I型(USH1)和粘多糖累积症I-H(MPS I-H) | 较高的生物相容性、剂量依赖性,可通过血脑屏障,无明显副作用 |
| 非氨基糖苷类[32] | | | | |
| PTC124 | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、粘多糖累积症VI型(MPS VI)、神经元蜡样脂褐质沉积症(NCLs)、贝敦氏病(Batten disease)、色素性视网膜炎(RP)、Usher综合征I型(USH1)和先天性无虹膜(Congenital aniridia) | 耐受性良好,无严重副作用 |
RTC13、RTC14和
RTC229 | 细胞 | 无 | 杜氏肌营养不良(DMD)和共济失调性毛细血管扩张症(AT) | 对3种提前终止密码子均有效 |
2.1 氨基糖苷类 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo
2
2009
... Current research status of drugs
Table 1 | 通读药物 | 实验模型 | 临床试验 | 相关疾病 | 特点与问题 |
| 氨基糖苷类 | | | | |
| 庆大霉素[27] | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、法布里病(Fabry)、尼曼匹克症A/B型(NPA/NPB)、全身性神经节苷脂贮积(Gangliosidosis I )、粘多糖累积症II型(MPS II) 、MPS I-H,MPS IIIB,MPS VI、血友病A和B(HA和HB)、无脉络膜症(Choroideremia)、先天性黑朦(LCA type 2)、眼缺损(Ocular coloboma )和Usher综合征I型(USH1) | 副作用如耳毒性、肾毒性和视网膜毒性 |
| 阿米卡星[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、粘多糖
累积症I-H(MPS I-H)和脊髓型肌萎缩症I型
(SMA I) | |
| 妥布霉素[29] | 动物 | 无 | 囊性纤维化和脊髓型肌萎缩症I型(SMA I) | |
| 巴龙霉素[30] | 细胞 | 无 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、
瑞特综合征(RTT)、无脉络膜症(Choroideremia)、粘多糖累积症I-H(MPS I-H)、眼缺损(Ocular
coloboma )和Usher综合征I型(USH1) | |
| G418[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、粘多糖累积症I-H(MPS I-H)、脊髓型肌萎缩症I型(SMA I)、先天性黑朦(LCA type 2)、色素性视网膜炎(RP)和Usher综合征I型(USH1) | |
| 氨基糖苷类衍生物 | | | | |
新霉素衍生物
(TC003和TC007)[29] | 动物和细胞 | 无 | 脊髓型肌萎缩症I型(SMA I) | 生物相容性更好 |
卡那霉素衍生物
(JL023)[29] | | | 脊髓型肌萎缩症I型(SMA I) | 无明显副作用 |
巴龙霉素衍生物
(NB30、NB54、
NB84和NB124)[31] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT) Usher综合征I型(USH1)和粘多糖累积症I-H(MPS I-H) | 较高的生物相容性、剂量依赖性,可通过血脑屏障,无明显副作用 |
| 非氨基糖苷类[32] | | | | |
| PTC124 | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、粘多糖累积症VI型(MPS VI)、神经元蜡样脂褐质沉积症(NCLs)、贝敦氏病(Batten disease)、色素性视网膜炎(RP)、Usher综合征I型(USH1)和先天性无虹膜(Congenital aniridia) | 耐受性良好,无严重副作用 |
RTC13、RTC14和
RTC229 | 细胞 | 无 | 杜氏肌营养不良(DMD)和共济失调性毛细血管扩张症(AT) | 对3种提前终止密码子均有效 |
2.1 氨基糖苷类 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
... [
28]
动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、粘多糖累积症I-H(MPS I-H)、脊髓型肌萎缩症I型(SMA I)、先天性黑朦(LCA type 2)、色素性视网膜炎(RP)和Usher综合征I型(USH1) | | | 氨基糖苷类衍生物 | | | | |
新霉素衍生物
(TC003和TC007)[29] | 动物和细胞 | 无 | 脊髓型肌萎缩症I型(SMA I) | 生物相容性更好 |
卡那霉素衍生物
(JL023)[29] | | | 脊髓型肌萎缩症I型(SMA I) | 无明显副作用 |
巴龙霉素衍生物
(NB30、NB54、
NB84和NB124)[31] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT) Usher综合征I型(USH1)和粘多糖累积症I-H(MPS I-H) | 较高的生物相容性、剂量依赖性,可通过血脑屏障,无明显副作用 |
| 非氨基糖苷类[32] | | | | |
| PTC124 | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、粘多糖累积症VI型(MPS VI)、神经元蜡样脂褐质沉积症(NCLs)、贝敦氏病(Batten disease)、色素性视网膜炎(RP)、Usher综合征I型(USH1)和先天性无虹膜(Congenital aniridia) | 耐受性良好,无严重副作用 |
RTC13、RTC14和
RTC229 | 细胞 | 无 | 杜氏肌营养不良(DMD)和共济失调性毛细血管扩张症(AT) | 对3种提前终止密码子均有效 |
2.1 氨基糖苷类 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts
3
2006
... Current research status of drugs
Table 1 | 通读药物 | 实验模型 | 临床试验 | 相关疾病 | 特点与问题 |
| 氨基糖苷类 | | | | |
| 庆大霉素[27] | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、法布里病(Fabry)、尼曼匹克症A/B型(NPA/NPB)、全身性神经节苷脂贮积(Gangliosidosis I )、粘多糖累积症II型(MPS II) 、MPS I-H,MPS IIIB,MPS VI、血友病A和B(HA和HB)、无脉络膜症(Choroideremia)、先天性黑朦(LCA type 2)、眼缺损(Ocular coloboma )和Usher综合征I型(USH1) | 副作用如耳毒性、肾毒性和视网膜毒性 |
| 阿米卡星[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、粘多糖
累积症I-H(MPS I-H)和脊髓型肌萎缩症I型
(SMA I) | |
| 妥布霉素[29] | 动物 | 无 | 囊性纤维化和脊髓型肌萎缩症I型(SMA I) | |
| 巴龙霉素[30] | 细胞 | 无 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、
瑞特综合征(RTT)、无脉络膜症(Choroideremia)、粘多糖累积症I-H(MPS I-H)、眼缺损(Ocular
coloboma )和Usher综合征I型(USH1) | |
| G418[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、粘多糖累积症I-H(MPS I-H)、脊髓型肌萎缩症I型(SMA I)、先天性黑朦(LCA type 2)、色素性视网膜炎(RP)和Usher综合征I型(USH1) | |
| 氨基糖苷类衍生物 | | | | |
新霉素衍生物
(TC003和TC007)[29] | 动物和细胞 | 无 | 脊髓型肌萎缩症I型(SMA I) | 生物相容性更好 |
卡那霉素衍生物
(JL023)[29] | | | 脊髓型肌萎缩症I型(SMA I) | 无明显副作用 |
巴龙霉素衍生物
(NB30、NB54、
NB84和NB124)[31] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT) Usher综合征I型(USH1)和粘多糖累积症I-H(MPS I-H) | 较高的生物相容性、剂量依赖性,可通过血脑屏障,无明显副作用 |
| 非氨基糖苷类[32] | | | | |
| PTC124 | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、粘多糖累积症VI型(MPS VI)、神经元蜡样脂褐质沉积症(NCLs)、贝敦氏病(Batten disease)、色素性视网膜炎(RP)、Usher综合征I型(USH1)和先天性无虹膜(Congenital aniridia) | 耐受性良好,无严重副作用 |
RTC13、RTC14和
RTC229 | 细胞 | 无 | 杜氏肌营养不良(DMD)和共济失调性毛细血管扩张症(AT) | 对3种提前终止密码子均有效 |
2.1 氨基糖苷类 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
... [
29]
动物和细胞 | 无 | 脊髓型肌萎缩症I型(SMA I) | 生物相容性更好 | 卡那霉素衍生物
(JL023)[29] | | | 脊髓型肌萎缩症I型(SMA I) | 无明显副作用 |
巴龙霉素衍生物
(NB30、NB54、
NB84和NB124)[31] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT) Usher综合征I型(USH1)和粘多糖累积症I-H(MPS I-H) | 较高的生物相容性、剂量依赖性,可通过血脑屏障,无明显副作用 |
| 非氨基糖苷类[32] | | | | |
| PTC124 | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、粘多糖累积症VI型(MPS VI)、神经元蜡样脂褐质沉积症(NCLs)、贝敦氏病(Batten disease)、色素性视网膜炎(RP)、Usher综合征I型(USH1)和先天性无虹膜(Congenital aniridia) | 耐受性良好,无严重副作用 |
RTC13、RTC14和
RTC229 | 细胞 | 无 | 杜氏肌营养不良(DMD)和共济失调性毛细血管扩张症(AT) | 对3种提前终止密码子均有效 |
2.1 氨基糖苷类 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
... [
29]
| | 脊髓型肌萎缩症I型(SMA I) | 无明显副作用 | 巴龙霉素衍生物
(NB30、NB54、
NB84和NB124)[31] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT) Usher综合征I型(USH1)和粘多糖累积症I-H(MPS I-H) | 较高的生物相容性、剂量依赖性,可通过血脑屏障,无明显副作用 |
| 非氨基糖苷类[32] | | | | |
| PTC124 | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、粘多糖累积症VI型(MPS VI)、神经元蜡样脂褐质沉积症(NCLs)、贝敦氏病(Batten disease)、色素性视网膜炎(RP)、Usher综合征I型(USH1)和先天性无虹膜(Congenital aniridia) | 耐受性良好,无严重副作用 |
RTC13、RTC14和
RTC229 | 细胞 | 无 | 杜氏肌营养不良(DMD)和共济失调性毛细血管扩张症(AT) | 对3种提前终止密码子均有效 |
2.1 氨基糖苷类 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
Translational bypass of nonsense mutations in zebrafish rep1, pax2.1 and lamb1 highlights a viable therapeutic option for untreatable genetic eye disease
1
2008
... Current research status of drugs
Table 1 | 通读药物 | 实验模型 | 临床试验 | 相关疾病 | 特点与问题 |
| 氨基糖苷类 | | | | |
| 庆大霉素[27] | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、法布里病(Fabry)、尼曼匹克症A/B型(NPA/NPB)、全身性神经节苷脂贮积(Gangliosidosis I )、粘多糖累积症II型(MPS II) 、MPS I-H,MPS IIIB,MPS VI、血友病A和B(HA和HB)、无脉络膜症(Choroideremia)、先天性黑朦(LCA type 2)、眼缺损(Ocular coloboma )和Usher综合征I型(USH1) | 副作用如耳毒性、肾毒性和视网膜毒性 |
| 阿米卡星[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、粘多糖
累积症I-H(MPS I-H)和脊髓型肌萎缩症I型
(SMA I) | |
| 妥布霉素[29] | 动物 | 无 | 囊性纤维化和脊髓型肌萎缩症I型(SMA I) | |
| 巴龙霉素[30] | 细胞 | 无 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、
瑞特综合征(RTT)、无脉络膜症(Choroideremia)、粘多糖累积症I-H(MPS I-H)、眼缺损(Ocular
coloboma )和Usher综合征I型(USH1) | |
| G418[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、粘多糖累积症I-H(MPS I-H)、脊髓型肌萎缩症I型(SMA I)、先天性黑朦(LCA type 2)、色素性视网膜炎(RP)和Usher综合征I型(USH1) | |
| 氨基糖苷类衍生物 | | | | |
新霉素衍生物
(TC003和TC007)[29] | 动物和细胞 | 无 | 脊髓型肌萎缩症I型(SMA I) | 生物相容性更好 |
卡那霉素衍生物
(JL023)[29] | | | 脊髓型肌萎缩症I型(SMA I) | 无明显副作用 |
巴龙霉素衍生物
(NB30、NB54、
NB84和NB124)[31] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT) Usher综合征I型(USH1)和粘多糖累积症I-H(MPS I-H) | 较高的生物相容性、剂量依赖性,可通过血脑屏障,无明显副作用 |
| 非氨基糖苷类[32] | | | | |
| PTC124 | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、粘多糖累积症VI型(MPS VI)、神经元蜡样脂褐质沉积症(NCLs)、贝敦氏病(Batten disease)、色素性视网膜炎(RP)、Usher综合征I型(USH1)和先天性无虹膜(Congenital aniridia) | 耐受性良好,无严重副作用 |
RTC13、RTC14和
RTC229 | 细胞 | 无 | 杜氏肌营养不良(DMD)和共济失调性毛细血管扩张症(AT) | 对3种提前终止密码子均有效 |
2.1 氨基糖苷类 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse
2
2012
... Current research status of drugs
Table 1 | 通读药物 | 实验模型 | 临床试验 | 相关疾病 | 特点与问题 |
| 氨基糖苷类 | | | | |
| 庆大霉素[27] | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、法布里病(Fabry)、尼曼匹克症A/B型(NPA/NPB)、全身性神经节苷脂贮积(Gangliosidosis I )、粘多糖累积症II型(MPS II) 、MPS I-H,MPS IIIB,MPS VI、血友病A和B(HA和HB)、无脉络膜症(Choroideremia)、先天性黑朦(LCA type 2)、眼缺损(Ocular coloboma )和Usher综合征I型(USH1) | 副作用如耳毒性、肾毒性和视网膜毒性 |
| 阿米卡星[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、粘多糖
累积症I-H(MPS I-H)和脊髓型肌萎缩症I型
(SMA I) | |
| 妥布霉素[29] | 动物 | 无 | 囊性纤维化和脊髓型肌萎缩症I型(SMA I) | |
| 巴龙霉素[30] | 细胞 | 无 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、
瑞特综合征(RTT)、无脉络膜症(Choroideremia)、粘多糖累积症I-H(MPS I-H)、眼缺损(Ocular
coloboma )和Usher综合征I型(USH1) | |
| G418[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、粘多糖累积症I-H(MPS I-H)、脊髓型肌萎缩症I型(SMA I)、先天性黑朦(LCA type 2)、色素性视网膜炎(RP)和Usher综合征I型(USH1) | |
| 氨基糖苷类衍生物 | | | | |
新霉素衍生物
(TC003和TC007)[29] | 动物和细胞 | 无 | 脊髓型肌萎缩症I型(SMA I) | 生物相容性更好 |
卡那霉素衍生物
(JL023)[29] | | | 脊髓型肌萎缩症I型(SMA I) | 无明显副作用 |
巴龙霉素衍生物
(NB30、NB54、
NB84和NB124)[31] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT) Usher综合征I型(USH1)和粘多糖累积症I-H(MPS I-H) | 较高的生物相容性、剂量依赖性,可通过血脑屏障,无明显副作用 |
| 非氨基糖苷类[32] | | | | |
| PTC124 | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、粘多糖累积症VI型(MPS VI)、神经元蜡样脂褐质沉积症(NCLs)、贝敦氏病(Batten disease)、色素性视网膜炎(RP)、Usher综合征I型(USH1)和先天性无虹膜(Congenital aniridia) | 耐受性良好,无严重副作用 |
RTC13、RTC14和
RTC229 | 细胞 | 无 | 杜氏肌营养不良(DMD)和共济失调性毛细血管扩张症(AT) | 对3种提前终止密码子均有效 |
2.1 氨基糖苷类 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
... Hurler综合征被认为是最严重的遗传性溶酶体贮积病,为常染色体隐性遗传病.由于IDUA基因变异导致所编码的α-L-艾杜糖醛酸苷酶缺乏,溶酶体内的粘多糖(GAG)不能正常降解,引发粘多糖贮积病.IDUA基因位于1号染色体,p.Gln70Ter和p.Trp402Ter是欧洲裔Hurler综合征患者中较常见的两种无义变异[36].2001年,Keeling等[37]使用Hurler综合征成纤维细胞系研究了庆大霉素对IDUA基因p.Gln70Ter和p.Trp402Ter变异的抑制活性,发现经庆大霉素处理后可以显著提升α-L-艾杜糖醛酸苷酶的活性.但是由于长期应用庆大霉素产生副作用,研究者也尝试设计更为有效且毒性更小的氨基糖苷类药物,其中一种氨基糖苷类衍生物-NB84,在疗效上可恢复α-L-艾杜糖醛酸苷酶活性,且几乎没有毒性[31]. ...
Translational read-through as an alternative approach for ocular gene therapy of retinal dystrophies caused by in-frame nonsense mutations
1
2014
... Current research status of drugs
Table 1 | 通读药物 | 实验模型 | 临床试验 | 相关疾病 | 特点与问题 |
| 氨基糖苷类 | | | | |
| 庆大霉素[27] | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、法布里病(Fabry)、尼曼匹克症A/B型(NPA/NPB)、全身性神经节苷脂贮积(Gangliosidosis I )、粘多糖累积症II型(MPS II) 、MPS I-H,MPS IIIB,MPS VI、血友病A和B(HA和HB)、无脉络膜症(Choroideremia)、先天性黑朦(LCA type 2)、眼缺损(Ocular coloboma )和Usher综合征I型(USH1) | 副作用如耳毒性、肾毒性和视网膜毒性 |
| 阿米卡星[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、粘多糖
累积症I-H(MPS I-H)和脊髓型肌萎缩症I型
(SMA I) | |
| 妥布霉素[29] | 动物 | 无 | 囊性纤维化和脊髓型肌萎缩症I型(SMA I) | |
| 巴龙霉素[30] | 细胞 | 无 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、
瑞特综合征(RTT)、无脉络膜症(Choroideremia)、粘多糖累积症I-H(MPS I-H)、眼缺损(Ocular
coloboma )和Usher综合征I型(USH1) | |
| G418[28] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT)、共济失调性毛细血管扩张症(AT)、粘多糖累积症I-H(MPS I-H)、脊髓型肌萎缩症I型(SMA I)、先天性黑朦(LCA type 2)、色素性视网膜炎(RP)和Usher综合征I型(USH1) | |
| 氨基糖苷类衍生物 | | | | |
新霉素衍生物
(TC003和TC007)[29] | 动物和细胞 | 无 | 脊髓型肌萎缩症I型(SMA I) | 生物相容性更好 |
卡那霉素衍生物
(JL023)[29] | | | 脊髓型肌萎缩症I型(SMA I) | 无明显副作用 |
巴龙霉素衍生物
(NB30、NB54、
NB84和NB124)[31] | 动物和细胞 | 无 | 囊性纤维化(CF)、瑞特综合征(RTT) Usher综合征I型(USH1)和粘多糖累积症I-H(MPS I-H) | 较高的生物相容性、剂量依赖性,可通过血脑屏障,无明显副作用 |
| 非氨基糖苷类[32] | | | | |
| PTC124 | 动物和患者 | 有 | 囊性纤维化(CF)、杜氏肌营养不良(DMD)、粘多糖累积症VI型(MPS VI)、神经元蜡样脂褐质沉积症(NCLs)、贝敦氏病(Batten disease)、色素性视网膜炎(RP)、Usher综合征I型(USH1)和先天性无虹膜(Congenital aniridia) | 耐受性良好,无严重副作用 |
RTC13、RTC14和
RTC229 | 细胞 | 无 | 杜氏肌营养不良(DMD)和共济失调性毛细血管扩张症(AT) | 对3种提前终止密码子均有效 |
2.1 氨基糖苷类 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
Binding of aminoglycoside antibiotics to helix 69 of 23S rRNA
1
2010
... 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
Fidelity of the eukaryotic codon-anticodon interaction: interference by aminoglycoside antibiotics
1
1984
... 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
Pharmaceutical therapies to recode nonsense mutations in inherited diseases
1
2012
... 氨基糖苷类是应用较广的一类抗生素,可通过干扰核糖体校正作用减少核糖体翻译过程的提前终止.氨基糖苷类药物选择性地与核糖体的解码中心结合,导致氨基酸的随机引入,从而克服肽链中的PTC,发生通读[33].例如,庆大霉素是一种常见的氨基糖苷类抗生素,通过与真核核糖体结合发挥无义抑制活性[34],选择性地在PTC中引入氨基酸,但不影响正常终止密码子.在作用过程中,庆大霉素通过促进近同工tRNA与核糖体A位点的结合而抑制校对过程,使翻译的保真度降低,从而导致误读和/或PTC抑制[35]. ...
Mucopolysaccharidosis type I: identification of 8 novel mutations and determination of the frequency of the two common α-L-iduronidase mutations (W402X and Q70X) among European patients
1
1994
... Hurler综合征被认为是最严重的遗传性溶酶体贮积病,为常染色体隐性遗传病.由于IDUA基因变异导致所编码的α-L-艾杜糖醛酸苷酶缺乏,溶酶体内的粘多糖(GAG)不能正常降解,引发粘多糖贮积病.IDUA基因位于1号染色体,p.Gln70Ter和p.Trp402Ter是欧洲裔Hurler综合征患者中较常见的两种无义变异[36].2001年,Keeling等[37]使用Hurler综合征成纤维细胞系研究了庆大霉素对IDUA基因p.Gln70Ter和p.Trp402Ter变异的抑制活性,发现经庆大霉素处理后可以显著提升α-L-艾杜糖醛酸苷酶的活性.但是由于长期应用庆大霉素产生副作用,研究者也尝试设计更为有效且毒性更小的氨基糖苷类药物,其中一种氨基糖苷类衍生物-NB84,在疗效上可恢复α-L-艾杜糖醛酸苷酶活性,且几乎没有毒性[31]. ...
Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of α-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation
1
2001
... Hurler综合征被认为是最严重的遗传性溶酶体贮积病,为常染色体隐性遗传病.由于IDUA基因变异导致所编码的α-L-艾杜糖醛酸苷酶缺乏,溶酶体内的粘多糖(GAG)不能正常降解,引发粘多糖贮积病.IDUA基因位于1号染色体,p.Gln70Ter和p.Trp402Ter是欧洲裔Hurler综合征患者中较常见的两种无义变异[36].2001年,Keeling等[37]使用Hurler综合征成纤维细胞系研究了庆大霉素对IDUA基因p.Gln70Ter和p.Trp402Ter变异的抑制活性,发现经庆大霉素处理后可以显著提升α-L-艾杜糖醛酸苷酶的活性.但是由于长期应用庆大霉素产生副作用,研究者也尝试设计更为有效且毒性更小的氨基糖苷类药物,其中一种氨基糖苷类衍生物-NB84,在疗效上可恢复α-L-艾杜糖醛酸苷酶活性,且几乎没有毒性[31]. ...
PTC124 targets genetic disorders caused by nonsense mutations
1
2007
... Ataluren (原称PTC124)是一种非氨基糖苷类的小分子化合物,能够在mRNA转录本的PTC位置随机插入氨基酸,诱导PTC通读,而不影响正常的终止密码子[38].DMD由编码抗肌萎缩蛋白的基因突变引起,患者主要表现为进行性近端肌肉无力,导致行走困难、依赖轮椅以及最终的呼吸和心脏衰竭恶化.由无义变异产生PTC导致截短蛋白或蛋白质功能丧失约占DMD病例的13%.Ataluren是通过药物的高通量筛选和化学优化而被发现的,它可以诱导核糖体读取PTC,而不是正常的终止密码子.当在无义突变的DMD小鼠模型中测试时,Ataluren可产生全长的功能性抗肌萎缩蛋白.在随后的人DMD实验也成功诱导出了全长功能性抗肌萎缩蛋白[39].基于PTC124药物的有效性,2017年PTC124通过了欧盟的年度审核.目前,PTC公司正在进行一个随机双盲、安慰剂对照、国际多中心的III期临床试验,以进一步证实PTC124长期治疗无义变异的DMD患者的效果和安全性.该临床试验总目标入组人数250人,中国入组人数50人,预期2021年将公布结果(https://clinicaltrials.gov; http://www.chinadrugtrials.org.cn). ...
Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: aminoglycosides and ataluren (PTC124)
1
2010
... Ataluren (原称PTC124)是一种非氨基糖苷类的小分子化合物,能够在mRNA转录本的PTC位置随机插入氨基酸,诱导PTC通读,而不影响正常的终止密码子[38].DMD由编码抗肌萎缩蛋白的基因突变引起,患者主要表现为进行性近端肌肉无力,导致行走困难、依赖轮椅以及最终的呼吸和心脏衰竭恶化.由无义变异产生PTC导致截短蛋白或蛋白质功能丧失约占DMD病例的13%.Ataluren是通过药物的高通量筛选和化学优化而被发现的,它可以诱导核糖体读取PTC,而不是正常的终止密码子.当在无义突变的DMD小鼠模型中测试时,Ataluren可产生全长的功能性抗肌萎缩蛋白.在随后的人DMD实验也成功诱导出了全长功能性抗肌萎缩蛋白[39].基于PTC124药物的有效性,2017年PTC124通过了欧盟的年度审核.目前,PTC公司正在进行一个随机双盲、安慰剂对照、国际多中心的III期临床试验,以进一步证实PTC124长期治疗无义变异的DMD患者的效果和安全性.该临床试验总目标入组人数250人,中国入组人数50人,预期2021年将公布结果(https://clinicaltrials.gov; http://www.chinadrugtrials.org.cn). ...
Germline E-cadherin mutations in hereditary diffuse gastric cancer: assessment of 42 new families and review of genetic screening criteria
1
2004
... 无义抑制子是一种tRNA的衍生物,其反密码子能够识别PTC,继而在PTC处掺入氨基酸,产生全长功能蛋白,防止翻译发生终止.抑制子介导的密码子-反密码子的相互作用,可以避免由于核糖体校正而发生的氨酰-tRNA排斥,因此可以有效抑制PTC.研究表明,抑制子tRNA可用于人体由无义变异引起的疾病的体细胞基因治疗.遗传性弥漫性胃癌(HDGC)是一种侵袭性、无法治愈的胃癌,主要由CDH1基因的无义变异引起,导致截短的E-钙粘蛋白产生[40].最近一项关于HDGC的研究显示,在给予抑制子tRNA诱导后,108个具有HDGC家族史的家庭中,有23个家庭(21.3%)的癌症的发病显著延迟,并使由携带PTC的CDH1基因可以编码产生大约30%功能性E-钙粘蛋白[41]. ...
Rescue of wild-type E-cadherin expression from nonsense-mutated cancer cells by a suppressor-tRNA
1
2014
... 无义抑制子是一种tRNA的衍生物,其反密码子能够识别PTC,继而在PTC处掺入氨基酸,产生全长功能蛋白,防止翻译发生终止.抑制子介导的密码子-反密码子的相互作用,可以避免由于核糖体校正而发生的氨酰-tRNA排斥,因此可以有效抑制PTC.研究表明,抑制子tRNA可用于人体由无义变异引起的疾病的体细胞基因治疗.遗传性弥漫性胃癌(HDGC)是一种侵袭性、无法治愈的胃癌,主要由CDH1基因的无义变异引起,导致截短的E-钙粘蛋白产生[40].最近一项关于HDGC的研究显示,在给予抑制子tRNA诱导后,108个具有HDGC家族史的家庭中,有23个家庭(21.3%)的癌症的发病显著延迟,并使由携带PTC的CDH1基因可以编码产生大约30%功能性E-钙粘蛋白[41]. ...
无义突变与“遗传补偿效应”
1
2019
... 综上所述,NMD逃逸使得携带PTC的转录本仍可编码具有部分功能的蛋白质,缓解患者表型.最近,马志鹏等[42]通过引入PTC利用NMD途径构建敲除capn3a基因的斑马鱼突变体,但是却发现该突变体发育正常并未出现预期的异常表型;同时他们还发现该突变体中多个capn家族基因上调弥补capn3a基因的功能,推测发生这种现象是由于遗传补偿效应,并提出假设:无义突变mRNA通过提高同源基因的表达来实现补偿效应.经实验证实无义突变与核酸序列同源性是激活遗传补偿效应的两个必要条件.该研究阐释了PTC不导致表型的另一重要机制,也为PTC相关疾病的致病机制和药物治疗研究提供了新方向. ...
无义突变与“遗传补偿效应”
1
2019
... 综上所述,NMD逃逸使得携带PTC的转录本仍可编码具有部分功能的蛋白质,缓解患者表型.最近,马志鹏等[42]通过引入PTC利用NMD途径构建敲除capn3a基因的斑马鱼突变体,但是却发现该突变体发育正常并未出现预期的异常表型;同时他们还发现该突变体中多个capn家族基因上调弥补capn3a基因的功能,推测发生这种现象是由于遗传补偿效应,并提出假设:无义突变mRNA通过提高同源基因的表达来实现补偿效应.经实验证实无义突变与核酸序列同源性是激活遗传补偿效应的两个必要条件.该研究阐释了PTC不导致表型的另一重要机制,也为PTC相关疾病的致病机制和药物治疗研究提供了新方向. ...







{kind=link}
{kind=link}
{kind=link}
{kind=link}