
遗传 ›› 2024, Vol. 46 ›› Issue (7): 560-569.doi: 10.16288/j.yczz.24-096
胡玉龙1( ), 杨芳1, 陈彦潼1, 谌烁楷1, 闫煜博1, 张跃博1, 吴晓林2,3, 汪加明4, 何俊1(), 高宁1()
), 杨芳1, 陈彦潼1, 谌烁楷1, 闫煜博1, 张跃博1, 吴晓林2,3, 汪加明4, 何俊1(), 高宁1()
收稿日期:2024-04-08
修回日期:2024-05-24
出版日期:2024-07-20
发布日期:2024-06-03
通讯作者:
何俊,教授,博士生导师,研究方向:猪遗传育种。E-mail: hejun@hunau.edu.cn;高宁,助理研究员,研究方向:分子数量遗传与动物育种。E-mail: gaon@hunau.edu.cn
作者简介:胡玉龙,硕士研究生,专业方向:分子数量遗传与动物育种。E-mail: huyulong1024864@163.com
基金资助:
Yulong Hu1(), Fang Yang1, Yantong Chen1, Shuokai Shen1, Yubo Yan1, Yuebo Zhang1, Xiaolin Wu2,3, Jiaming Wang4, Jun He1(), Ning Gao1()
Received:2024-04-08
Revised:2024-05-24
Published:2024-07-20
Online:2024-06-03
Supported by:摘要:
基因组预测已成为畜禽、作物遗传评估和人类疾病风险预测的主要技术,但经典的基因组预测方法在性状遗传调控机制等生物学先验信息的整合方面有一定的不足。本研究提出一种将mRNA转录本信息整合应用于复杂性状表型预测的方法。基于国际上广泛应用于数量遗传学研究的果蝇群体,对本研究提出的新方法进行准确性评估。结果显示,整合mRNA转录本,可有效提高部分性状基因组预测准确性,但对部分性状的表型预测准确性没有改善。与GBLUP相比,雄性果蝇D-香芹酮嗅觉反应(dCarvone)准确性由0.256提高到0.274,提高幅度7%。雄性果蝇咖啡因耐受反应(cafe)准确性由0.355提高到0.401,提高幅度13%。雄性果蝇百草枯耐受反应(survival_paraquat)准确性由0.101提高到0.138,提高幅度36%。雌性果蝇1-已醇嗅觉反应(1hexanol)准确性由0.147提高到0.210,提高幅度43%。综上所述,对于部分性状,通过整合mRNA转录本可有效提高基因组预测准确性(提高幅度为7%~43%)。对于部分性状,整合mRNA转录本并考虑互作效应可进一步提高预测准确性。
胡玉龙, 杨芳, 陈彦潼, 谌烁楷, 闫煜博, 张跃博, 吴晓林, 汪加明, 何俊, 高宁. 整合mRNA转录本与基因组信息的基因组选择方法研究[J]. 遗传, 2024, 46(7): 560-569.
Yulong Hu, Fang Yang, Yantong Chen, Shuokai Shen, Yubo Yan, Yuebo Zhang, Xiaolin Wu, Jiaming Wang, Jun He, Ning Gao. Integrating mRNA transcripts and genomic information into genomic prediction[J]. Hereditas(Beijing), 2024, 46(7): 560-569.

图1
SNP与转录本映射关系 id:个体编号;Paternal:父源;Maternal:母源。"
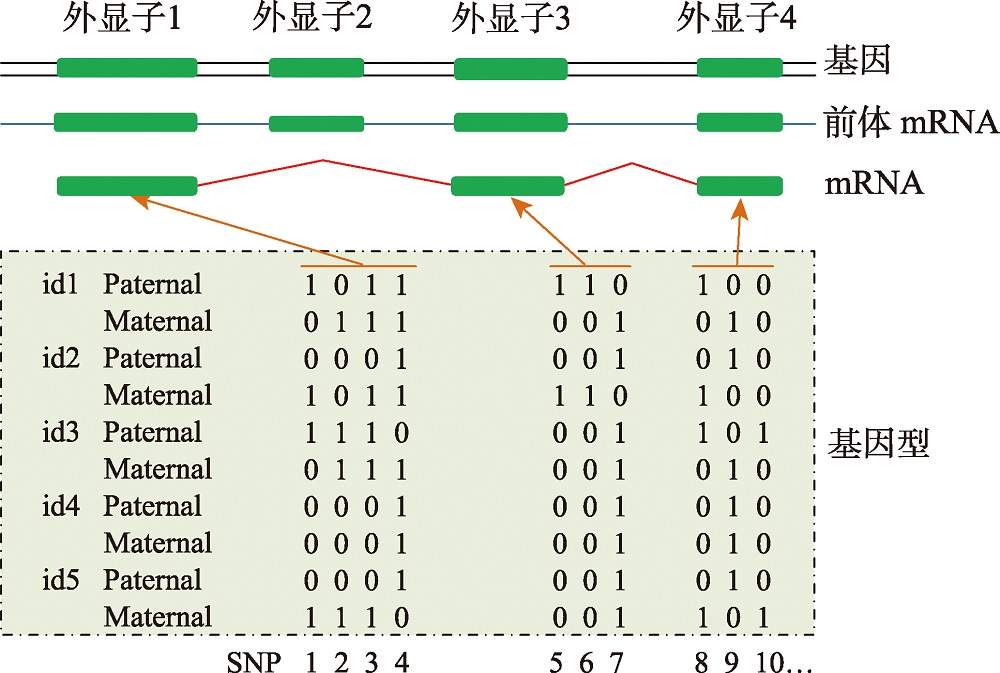

图2
基于转录本的单倍型构建策略 id:个体编号;Paternal:父源;Maternal:母源;hap:单倍型。"
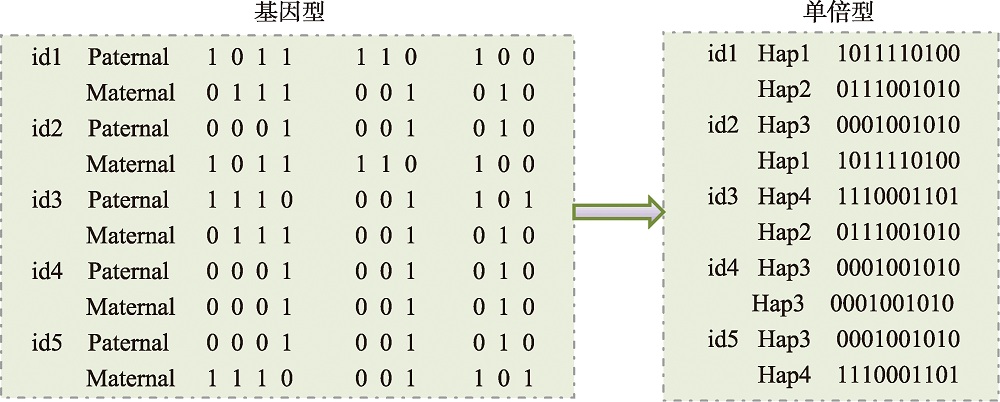

图3
预测模型中的单倍型编码策略 id:个体编号;hap:单倍型。"

表1
基因组预测模型基本描述"
| 模型 | 表达式 | 遗传效应 |
|---|---|---|
| GBLUP | ||
| GmBLUP | ||
| GHBLUP|T | ||
| CHM|T | ||
| CHE|T | ||
| GmBLUP* | ||
| GHBLUP|T* | ||
| CHM|T* | ||
| CHE|T* |
表2
DGPR2果蝇mRNA转录本的预测变量描述性统计"
| 项目 | 最小值 | 最大值 | 中位数 | 平均数 | 标准差 |
|---|---|---|---|---|---|
| 外显子数量 | 1 | 140 | 32 | 104 | 6.82 |
| CDS数量 | 1 | 116 | 3 | 4.55 | 4.57 |
| 转录本数量 | 1 | 75 | 2 | 2.23 | 2.31 |
| 转录本SNP数量 | 1 | 995 | 18 | 30.45 | 47.61 |
| 转录本等位基因数量 (质控前) | 2 | 195 | 37 | 49.74 | 42.83 |
| 转录本等位基因数量 (质控后) | 2 | 30 | 14 | 13.30 | 6.22 |
| 转录本基因型数量 (质控前) | 2 | 183 | 37 | 49.63 | 42.45 |
| 转录本基因型数量 (质控后) | 2 | 32 | 14 | 13.46 | 6.29 |
表3
不同基因组预测模型对果蝇多个性状基因组预测的准确性"
| 性状 | GBLUP | GmBLUP | GmBLUP* | GHBLUP|T | GHBLUP|T* | CHM|T | CHM|T* | CHE|T | CHE|T* |
|---|---|---|---|---|---|---|---|---|---|
| startle(F) | 0.347±0.009 | 0.361±0.009 | 0.358±0.010 | 0.355±0.009 | 0.341±0.009 | 0.364±0.009 | 0.357±0.009 | 0.321±0.009 | 0.343±0.009 |
| startle(M) | 0.315±0.010 | 0.329±0.010 | 0.327±0.010 | 0.324±0.010 | 0.314±0.010 | 0.321±0.010 | 0.313±0.009 | 0.284±0.010 | 0.310±0.010 |
| starvation(F) | 0.344±0.008 | 0.329±0.008 | 0.334±0.008 | 0.324±0.008 | 0.338±0.007 | 0.325±0.008 | 0.341±0.008 | 0.282±0.008 | 0.344±0.008 |
| starvation(M) | 0.362±0.007 | 0.346±0.007 | 0.351±0.007 | 0.361±0.006 | 0.353±0.006 | 0.350±0.007 | 0.360±0.007 | 0.299±0.007 | 0.362±0.007 |
| 1hexanol(F) | 0.147±0.009 | 0.152±0.010 | 0.136±0.010 | 0.194±0.010 | 0.189±0.010 | 0.210±0.010 | 0.208±0.010 | 0.206±0.009 | 0.199±0.010 |
| 1hexanol(M) | 0.198±0.010 | 0.181±0.010 | 0.190±0.010 | 0.203±0.009 | 0.192±0.009 | 0.202±0.009 | 0.189±0.010 | 0.205±0.009 | 0.197±0.009 |
| 2heptanone(F) | 0.284±0.007 | 0.270±0.007 | 0.277±0.007 | 0.261±0.007 | 0.284±0.007 | 0.228±0.007 | 0.285±0.007 | 0.247±0.006 | 0.284±0.007 |
| 2heptanone(M) | 0.137±0.009 | 0.140±0.010 | 0.126±0.008 | 0.131±0.009 | 0.125±0.009 | 0.106±0.009 | 0.132±0.009 | 0.134±0.010 | 0.124±0.010 |
| 2phenylEthylAlcohol(F) | 0.332±0.005 | 0.327±0.005 | 0.317±0.005 | 0.328±0.005 | 0.324±0.005 | 0.333±0.005 | 0.325±0.005 | 0.301±0.006 | 0.330±0.004 |
| 2phenylEthylAlcohol(M) | 0.143±0.008 | 0.140±0.008 | 0.126±0.008 | 0.132±0.009 | 0.128±0.009 | 0.123±0.009 | 0.133±0.010 | 0.114±0.010 | 0.128±0.010 |
| benz(F) | 0.291±0.011 | 0.302±0.010 | 0.298±0.010 | 0.280±0.010 | 0.280±0.010 | 0.299±0.010 | 0.285±0.010 | 0.266±0.011 | 0.288±0.011 |
| benz(M) | 0.198±0.014 | 0.210±0.013 | 0.205±0.013 | 0.182±0.015 | 0.192±0.015 | 0.205±0.014 | 0.192±0.014 | 0.161±0.017 | 0.195±0.015 |
| benzaldehyde(F) | 0.392±0.007 | 0.399±0.008 | 0.399±0.008 | 0.361±0.008 | 0.387±0.007 | 0.370±0.007 | 0.386±0.007 | 0.338±0.007 | 0.389±0.007 |
| benzaldehyde(M) | 0.307±0.008 | 0.318±0.007 | 0.313±0.007 | 0.288±0.008 | 0.299±0.008 | 0.293±0.007 | 0.300±0.008 | 0.251±0.006 | 0.303±0.008 |
| cafe(F) | 0.255±0.010 | 0.259±0.010 | 0.248±0.011 | 0.261±0.011 | 0.247±0.011 | 0.253±0.010 | 0.240±0.011 | 0.256±0.009 | 0.249±0.010 |
| cafe(M) | 0.355±0.007 | 0.372±0.007 | 0.370±0.007 | 0.401±0.007 | 0.400±0.007 | 0.401±0.007 | 0.401±0.007 | 0.378±0.006 | 0.349±0.008 |
| dCarvone(F) | 0.211±0.012 | 0.221±0.011 | 0.209±0.011 | 0.222±0.012 | 0.212±0.013 | 0.228±0.012 | 0.217±0.013 | 0.232±0.011 | 0.211±0.013 |
| dCarvone(M) | 0.256±0.011 | 0.241±0.011 | 0.238±0.011 | 0.274±0.011 | 0.268±0.011 | 0.256±0.011 | 0.241±0.011 | 0.242±0.011 | 0.252±0.010 |
| ethylAcetate(F) | 0.435±0.007 | 0.433±0.006 | 0.427±0.007 | 0.403±0.008 | 0.433±0.007 | 0.401±0.007 | 0.426±0.008 | 0.400±0.007 | 0.434±0.007 |
| ethylAcetate(M) | 0.336±0.014 | 0.338±0.014 | 0.329±0.014 | 0.311±0.015 | 0.327±0.014 | 0.314±0.014 | 0.328±0.014 | 0.279±0.015 | 0.334±0.014 |
| ethylButyrate(F) | 0.383±0.007 | 0.383±0.008 | 0.373±0.008 | 0.347±0.008 | 0.382±0.007 | 0.356±0.008 | 0.381±0.007 | 0.336±0.008 | 0.382±0.007 |
| ethylButyrate(M) | 0.463±0.007 | 0.468±0.007 | 0.456±0.007 | 0.432±0.008 | 0.460±0.007 | 0.422±0.008 | 0.460±0.007 | 0.391±0.008 | 0.460±0.007 |
| etoh_e1(F) | 0.185±0.007 | 0.177±0.008 | 0.168±0.008 | 0.158±0.008 | 0.178±0.008 | 0.174±0.009 | 0.167±0.008 | 0.179±0.009 | 0.172±0.008 |
| etoh_e1(M) | 0.068±0.009 | 0.060±0.010 | 0.063±0.009 | 0.033±0.010 | 0.062±0.009 | 0.059±0.011 | 0.051±0.010 | 0.041±0.009 | 0.059±0.009 |
| etoh_e2(F) | 0.197±0.010 | 0.189±0.009 | 0.183±0.010 | 0.160±0.010 | 0.189±0.010 | 0.179±0.009 | 0.177±0.010 | 0.171±0.011 | 0.175±0.012 |
| etoh_e2(M) | 0.058±0.010 | 0.047±0.010 | 0.047±0.010 | 0.059±0.009 | 0.048±0.009 | 0.070±0.009 | 0.052±0.009 | 0.063±0.008 | 0.053±0.008 |
| eugenol(F) | 0.235±0.012 | 0.244±0.012 | 0.233±0.013 | 0.201±0.014 | 0.217±0.012 | 0.180±0.013 | 0.228±0.013 | 0.191±0.015 | 0.228±0.012 |
| eugenol(M) | 0.176±0.009 | 0.205±0.009 | 0.200±0.008 | 0.151±0.010 | 0.153±0.010 | 0.144±0.009 | 0.165±0.010 | 0.107±0.012 | 0.169±0.009 |
| helional(F) | 0.136±0.014 | 0.146±0.015 | 0.135±0.015 | 0.100±0.015 | 0.111±0.014 | 0.085±0.015 | 0.125±0.014 | 0.106±0.013 | 0.112±0.013 |
| helional(M) | 0.101±0.012 | 0.062±0.013 | 0.092±0.013 | 0.056±0.014 | 0.088±0.013 | 0.031±0.014 | 0.092±0.012 | 0.061±0.014 | 0.091±0.012 |
| hexanal(F) | 0.310±0.008 | 0.303±0.007 | 0.301±0.008 | 0.310±0.008 | 0.303±0.008 | 0.298±0.008 | 0.306±0.008 | 0.274±0.008 | 0.306±0.008 |
| hexanal(M) | 0.209±0.007 | 0.214±0.007 | 0.198±0.007 | 0.224±0.009 | 0.209±0.007 | 0.215±0.009 | 0.200±0.007 | 0.200±0.009 | 0.202±0.008 |
| lCarvone(F) | 0.279±0.010 | 0.276±0.010 | 0.268±0.010 | 0.274±0.010 | 0.265±0.010 | 0.293±0.010 | 0.274±0.010 | 0.276±0.011 | 0.277±0.010 |
| lCarvone(M) | 0.341±0.007 | 0.284±0.007 | 0.350±0.007 | 0.312±0.007 | 0.349±0.007 | 0.312±0.006 | 0.350±0.007 | 0.296±0.008 | 0.349±0.007 |
| methylSalicylate(F) | 0.365±0.009 | 0.377±0.009 | 0.369±0.009 | 0.364±0.008 | 0.351±0.009 | 0.353±0.008 | 0.350±0.009 | 0.336±0.008 | 0.358±0.009 |
| methylSalicylate(M) | 0.337±0.007 | 0.336±0.008 | 0.320±0.007 | 0.308±0.008 | 0.335±0.007 | 0.293±0.009 | 0.336±0.007 | 0.276±0.008 | 0.336±0.007 |
| startle_msb_sensitivity(F) | 0.056±0.010 | 0.064±0.011 | 0.057±0.010 | 0.093±0.011 | 0.089±0.011 | 0.088±0.010 | 0.082±0.010 | 0.097±0.011 | 0.092±0.011 |
| startle_msb_sensitivity(M) | 0.006±0.014 | 0.015±0.013 | 0.006±0.014 | 0.082±0.014 | 0.070±0.013 | 0.082±0.014 | 0.071±0.013 | 0.078±0.015 | 0.066±0.014 |
| survival_paraquat(F) | 0.285±0.007 | 0.264±0.008 | 0.274±0.007 | 0.252±0.008 | 0.284±0.006 | 0.265±0.008 | 0.276±0.007 | 0.237±0.008 | 0.286±0.006 |
| survival_paraquat(M) | 0.101±0.009 | 0.108±0.009 | 0.087±0.009 | 0.115±0.011 | 0.106±0.010 | 0.138±0.010 | 0.129±0.010 | 0.091±0.012 | 0.084±0.010 |
| [1] |
Meuwissen TH, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics, 2001, 157(4): 1819-1829.
doi: 10.1093/genetics/157.4.1819 pmid: 11290733 |
| [2] |
Henderson CR. Best linear unbiased estimation and prediction under a selection model. Biometrics, 1975, 31(2): 423-447.
pmid: 1174616 |
| [3] |
Hayes BJ, Bowman PJ, Chamberlain AJ, Goddard ME. Invited review: genomic selection in dairy cattle: progress and challenges. J Dairy Sci, 2009, 92(2): 433-443.
doi: 10.3168/jds.2008-1646 pmid: 19164653 |
| [4] |
Zhao YX, Hou Y, Xu YY, Luan Y, Zhou HH, Qi XL, Hu MY, Wang DY, Wang ZX, Fu YH, Li JJ, Zhang SX, Chen JH, Han JL, Li XY, Zhao SH. A compendium and comparative epigenomics analysis of cis-regulatory elements in the pig genome. Nat Commun, 2021, 12(1): 2217.
doi: 10.1038/s41467-021-22448-x pmid: 33850120 |
| [5] | Tang ZS, Yin D, Yin LL, Ma YL, Xiang T, Zhu MJ, Yu M, Liu XL, Li XY, Qiu XT, Zhao SH. To evaluate the “two-step” genomic selection strategy in pig by simulation. Sci Agric Sin, 2021, 54(21): 4677-4686. |
|
唐振双, 殷东, 尹立林, 马云龙, 项韬, 朱猛进, 余梅, 刘小磊, 李新云, 邱小田, 赵书红. 猪基因组选择“两步走”策略的计算机模拟评估. 中国农业科学, 2021, 54(21): 4677-4686.
doi: 10.3864/j.issn.0578-1752.2021.21.016 |
|
| [6] |
VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci, 2008, 91(11): 4414-4423.
doi: 10.3168/jds.2007-0980 pmid: 18946147 |
| [7] |
Legarra A, Aguilar I, Misztal I. A relationship matrix including full pedigree and genomic information. J Dairy Sci, 2009, 92(9): 4656-4663.
doi: 10.3168/jds.2009-2061 pmid: 19700729 |
| [8] | Christensen OF, Lund MS. Genomic prediction when some animals are not genotyped. Genet Sel Evol, 2010, 42(1): 2. |
| [9] | Ye SP, Gao N, Zheng RR, Chen ZT, Teng JY, Yuan XL, Zhang H, Chen ZM, Zhang XQ, Li JQ, Zhang Z. Strategies for obtaining and pruning imputed whole- genome sequence data for genomic prediction. Front Genet, 2019, 10: 673. |
| [10] |
Al Kalaldeh M, Gibson J, Duijvesteijn N, Daetwyler HD, MacLeod I, Moghaddar N, Lee SH, van der Werf JHJ. Using imputed whole-genome sequence data to improve the accuracy of genomic prediction for parasite resistance in Australian sheep. Genet Sel Evol, 2019, 51(1): 32.
doi: 10.1186/s12711-019-0476-4 pmid: 31242855 |
| [11] |
Song HL, Ye SP, Jiang YF, Zhang Z, Zhang Q, Ding XD. Using imputation-based whole-genome sequencing data to improve the accuracy of genomic prediction for combined populations in pigs. Genet Sel Evol, 2019, 51(1): 58.
doi: 10.1186/s12711-019-0500-8 pmid: 31638889 |
| [12] | Zhang Z, Ober U, Erbe M, Zhang H, Gao N, He JL, Li JQ, Simianer H. Improving the accuracy of whole genome prediction for complex traits using the results of genome wide association studies. PLoS One, 2014, 9(3): e93017. |
| [13] |
Gao N, Martini JWR, Zhang Z, Yuan XL, Zhang H, Simianer H, Li JQ. Incorporating gene annotation into genomic prediction of complex phenotypes. Genetics, 2017, 207(2): 489-501.
doi: 10.1534/genetics.117.300198 pmid: 28839043 |
| [14] |
Gao N, Teng JY, Ye SP, Yuan XL, Huang SW, Zhang H, Zhang XQ, Li JQ, Zhang Z. Genomic prediction of complex phenotypes using genic similarity based relatedness matrix. Front Genet, 2018, 9: 364.
doi: 10.3389/fgene.2018.00364 pmid: 30233646 |
| [15] |
Edwards SM, Sørensen IF, Sarup P, Mackay TFC, Sørensen P. Genomic prediction for quantitative traits is improved by mapping variants to gene ontology categories in Drosophila melanogaster. Genetics, 2016, 203(4): 1871-1883.
doi: 10.1534/genetics.116.187161 pmid: 27235308 |
| [16] | Christensen OF, Börner V, Varona L, Legarra A. Genetic evaluation including intermediate omics features. Genetics, 2021, 219(2): iyab130. |
| [17] | Hu ZL, Park CA, Reecy JM. Developmental progress and current status of the animal QTLdb. Nucleic Acids Res, 2016, 44(D1): D827-D833. |
| [18] | Zhang Z, Erbe M, He JL, Ober U, Gao N, Zhang H, Simianer H, Li JQ. Accuracy of whole-genome prediction using a genetic architecture-enhanced variance-covariance matrix. G3 (Bethesda), 2015, 5(4): 615-627. |
| [19] | de Los Campos G, Vazquez AI, Fernando R, Klimentidis YC, Sorensen D. Prediction of complex human traits using the genomic best linear unbiased predictor. PLoS Genet, 2013, 9(7): e1003608. |
| [20] | Ramstein GP, Evans J, Kaeppler SM, Mitchell RB, Vogel KP, Buell CR, Casler MD. Accuracy of genomic prediction in switchgrass (Panicum virgatum L.) improved by accounting for linkage disequilibrium. G3 (Bethesda), 2016, 6(4): 1049-1062. |
| [21] | Huang W, Richards S, Carbone MA, Zhu DH, Anholt RRH, Ayroles JF, Duncan L, Jordan KW, Lawrence F, Magwire MM, Warner CB, Blankenburg K, Han Y, Javaid M, Jayaseelan J, Jhangiani SN, Muzny D, Ongeri F, Perales L, Wu YQ, Zhang YQ, Zou XY, Stone EA, Gibbs RA, Mackay TFC. Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proc Nat Acad Sci USA, 2012, 109(39): 15553-15559. |
| [22] | Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu DH, Casillas S, Han Y, Magwire MM, Cridland JM, Richardson MF, Anholt RRH, Barrón M, Bess C, Blankenburg KP, Carbone MA, Castellano D, Chaboub L, Duncan L, Harris Z, Javaid M, Jayaseelan JC, Jhangiani SN, Jordan KW, Lara F, Lawrence F, Lee SL, Librado P, Linheiro RS, Lyman RF, Mackey AJ, Munidasa M, Muzny DM, Nazareth L, Newsham I, Perales L, Pu LL, Qu C, Ràmia M, Reid JG, Rollmann SM, Rozas J, Saada N, Turlapati L, Worley KC, Wu YQ, Yamamoto A, Zhu YM, Bergman CM, Thornton KR, Mittelman D, Gibbs RA. The Drosophila melanogaster genetic reference panel. Nature, 2012, 482(7384): 173-178. |
| [23] |
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 2007, 81(3): 559-575.
doi: 10.1086/519795 pmid: 17701901 |
| [24] |
Browning BL, Zhou Y, Browning SR. A one-penny imputed genome from next-generation reference panels. Am J Hum Genet, 2018, 103(3): 338-348.
doi: S0002-9297(18)30242-8 pmid: 30100085 |
| [25] |
Browning BL, Tian XW, Zhou Y, Browning SR. Fast two-stage phasing of large-scale sequence data. Am J Hum Genet, 2021, 108(10): 1880-1890.
doi: 10.1016/j.ajhg.2021.08.005 pmid: 34478634 |
| [26] |
Martini JWR, Wimmer V, Erbe M, Simianer H. Epistasis and covariance: how gene interaction translates into genomic relationship. Theor Appl Genet, 2016, 129(5): 963-976.
doi: 10.1007/s00122-016-2675-5 pmid: 26883048 |
| [27] |
Li ZC, Gao N, Martini JWR, Simianer H. Integrating gene expression data into genomic prediction. Front Genet, 2019, 10: 126.
doi: 10.3389/fgene.2019.00126 pmid: 30858865 |
| [28] |
Hickey JM, Kinghorn BP, Tier B, Clark SA, van der Werf JHJ, Gorjanc G. Genomic evaluations using similarity between haplotypes. J Anim Breed Genet, 2013, 130(4): 259-269.
doi: 10.1111/jbg.12020 pmid: 23855628 |
| [29] |
Dong C, Wang SJ, Zhao ZJ, Ji X, Shen Q, Yu Y, Cui SD, Wang JG, Chen ZY, Wang JY, Guo ZY, Wu PX, Tang GQ. Genomic prediction of pig growth traits based on machine learning. Hereditas(Beijing), 2023, 45(10): 922-932.
doi: 10.16288/j.yczz.23-120 pmid: 37872114 |
| 陈栋, 王书杰, 赵真坚, 姬祥, 申琦, 余杨, 崔晟頔, 王俊戈, 陈子旸, 王金勇, 郭宗义, 吴平先, 唐国庆. 基于机器学习的猪生长性状基因组预测. 遗传, 2023, 45(10): 922-932. | |
| [30] | Azodi CB, Pardo J, VanBuren R, de Los Campos G, Shiu SH. Transcriptome-based prediction of complex traits in maize. Plant Cell, 2020, 32(1): 139-151. |
| [1] | 杨岸奇, 陈斌, 冉茂良, 杨广民, 曾诚. 基因组选择在猪杂交育种中的应用[J]. 遗传, 2020, 42(2): 145-152. |
| [2] | 何俊,Fernando B. Lopes,吴晓林. 动物基因组选配方法与应用[J]. 遗传, 2019, 41(6): 486-493. |
| [3] | 赵志达,张莉. 基因组选择在绵羊育种中的应用[J]. 遗传, 2019, 41(4): 293-303. |
| [4] | 王凤红,张磊,李晓凯,范一星,乔贤,龚高,严晓春,张令天,王志英,王瑞军,刘志红,王志新,何利兵,张燕军,李金泉,赵艳红,苏蕊. 山羊基因组研究进展[J]. 遗传, 2019, 41(10): 928-938. |
| [5] | 何俊,钱长嵩,RichardG.TaitJr.,StewartBauck,吴晓林. SNP芯片数据估计动物个体基因组品种构成的方法及应用[J]. 遗传, 2018, 40(4): 305-314. |
| [6] | 李宏伟,王瑞军,王志英,李学武,王振宇,张燕军,苏蕊,刘志红,李金泉. 家畜基因组选择研究进展[J]. 遗传, 2017, 39(5): 377-387. |
| [7] | 谈成, 边成, 杨达, 李宁, 吴珍芳, 胡晓湘, . 基因组选择技术在农业动物育种中的应用[J]. 遗传, 2017, 39(11): 1033-1045. |
| [8] | 王重龙, 丁向东, 刘剑锋, 殷宗俊, 张勤. 基因组育种值估计的贝叶斯方法研究进展[J]. 遗传, 2014, 36(2): 111-118. |
| [9] | 李恒德,包振民,孙效文. 基因组选择及其应用[J]. 遗传, 2011, 33(12): 1308-1316. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||

www.chinagene.cn
备案号:京ICP备09063187号-4
总访问:,今日访问:,当前在线:
