
遗传 ›› 2020, Vol. 42 ›› Issue (7): 691-702.doi: 10.16288/j.yczz.20-022
刘硕1, 曾志1, 曾凡才2, 杜萌泽2( )
)
收稿日期:2020-02-20
修回日期:2020-05-11
出版日期:2020-07-20
发布日期:2020-06-01
作者简介:刘硕,在读博士研究生,专业方向:微生物基因组学。E-mail: 基金资助:
Shuo Liu1, Zhi Zeng1, Fancai Zeng2, Mengze Du2()
Received:2020-02-20
Revised:2020-05-11
Published:2020-07-20
Online:2020-06-01
Supported by:摘要:
随着测序技术的不断发展,产生了海量的基因组测序数据,极大地丰富了公共遗传数据资源。同时为了应对大量基因组数据的产生,基因组比较和注释算法、工具不断更新,使得联合多种注释工具得到更准确的蛋白编码基因的注释信息成为可能。目前公共数据库的原核生物基因组测序和装配有些是10多年前的,存在大量预测的功能未知的编码基因。为了提升美国国家生物信息中心(National Center for Biotechnology Information, NCBI)数据库中基因组的注释质量,本研究联合使用多种原核基因识别算法/软件和基因表达数据重注释1587个细菌和古细菌基因组。首先,利用Z曲线的33个变量从177个基因组原注释中识别获得3092个被过度注释为蛋白编码基因的序列;其次,通过同源比对为939个基因组中的4447个功能未知的蛋白编码基因注释上具体功能;最后,通过联合采用ZCURVE 3.0和Glimmer 3.02以及Prodigal这3种高精度的、广泛使用且基于算法不同而互补的基因识别软件来寻找漏注释基因。最终,从9个基因组中找到了2003个被漏注释的蛋白编码基因,这些基因属于多个蛋白质直系同源簇(clusters of orthologous groups of proteins, COG)。本研究使用新的工具并结合多组学数据重新注释早期测序的细菌和古细菌基因组,不仅为新测序菌株提供注释方法参考,而且这些重注释后得到的细菌基因序列也会对后续基础研究有所帮助。
刘硕, 曾志, 曾凡才, 杜萌泽. 基于序列相似性和Z曲线方法重注释原核生物蛋白编码基因[J]. 遗传, 2020, 42(7): 691-702.
Shuo Liu, Zhi Zeng, Fancai Zeng, Mengze Du. Comprehensive re-annotation of protein-coding genes for prokaryotic genomes by Z-curve and similarity-based methods[J]. Hereditas(Beijing), 2020, 42(7): 691-702.
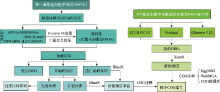
图1
重注释流程图"
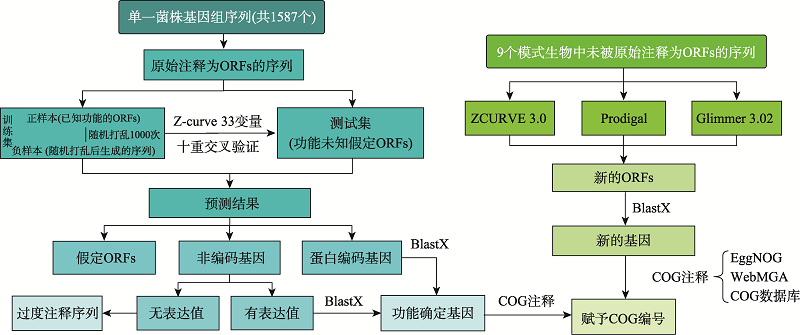
图2
1587个基因组的测序时间分布"
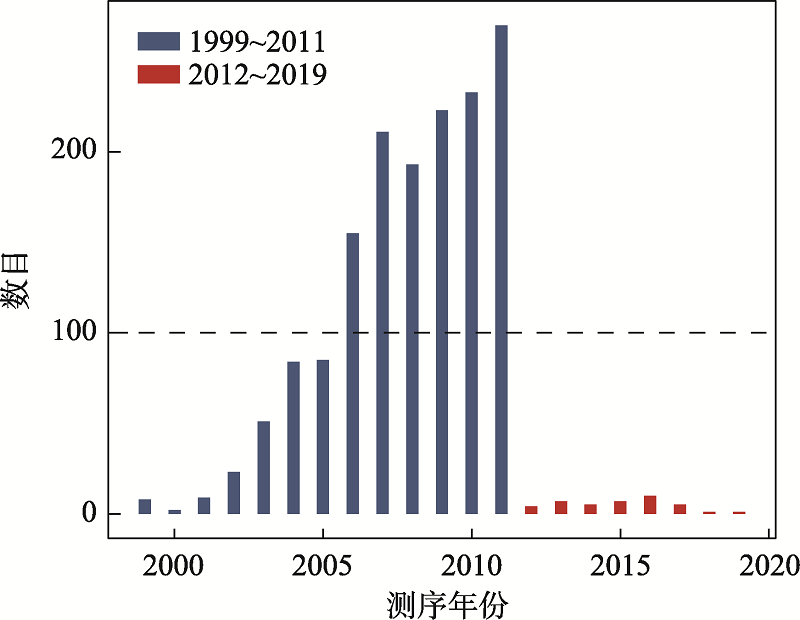
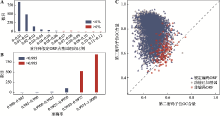
图3
假定ORFs的比率及不同准确率对应的基因组数量及大豆根瘤菌(B. japonicum USDA 110)3类序列两个密码子位GC含量 A:不同比率的重注释假定ORFs对应基因组的数目;B:不同识别非编码ORFs的准确率对应基因组的数目;C:大豆根瘤菌3类序列对应的第2和第3位密码子GC含量。"
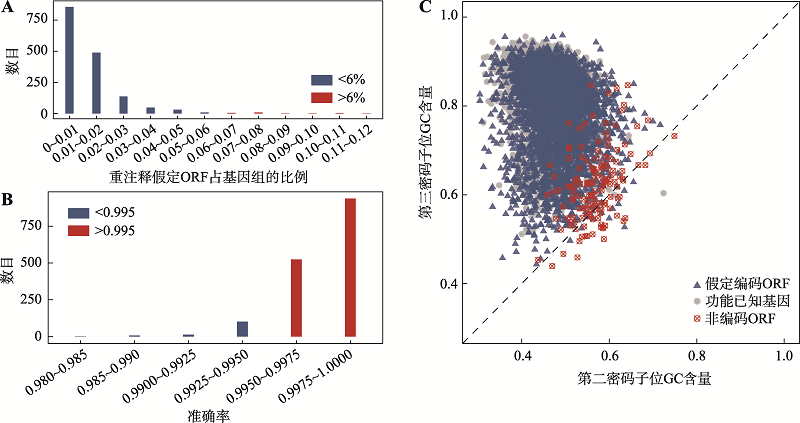
表1
识别出过度注释的ORFs多于20的菌株基因组的信息"
| 菌株 | NC序列号 | 数量 | 菌株 | NC序列号 | 数量 |
|---|---|---|---|---|---|
| 慢性型大豆根瘤菌(B. japonicum USDA 110) | NC_004463 | 147 | 金黄色葡萄球菌 (S. aureus subsp. Aureus MW2) | NC_003923 | 36 |
| 哈氏弧菌(Vibrio harveyi ATCC BAA-1116) | NC_009784 | 133 | 黑海甲烷袋状菌 (Methanoculleus marisnigri JR1) | NC_009051 | 34 |
| 大肠杆菌(E. coli CFT073) | NC_004431 | 119 | 双叶钩端螺旋体血清型Patoc菌株 (Leptospira biflexa serovar Patoc strain 'Patoc 1) | NC_010602 | 34 |
| 织片草螺菌 (Herbaspirillum seropedicae SmR1) | NC_014323 | 109 | 拟杆菌属 (Bacteroides salanitronis DSM 18170) | NC_015164 | 34 |
| 结核分枝杆菌 (Mycobacterium tuberculosis CDC1551) | NC_002755 | 98 | 巴尔通体杆菌(Bartonella clarridgeiae 73) | NC_014932 | 33 |
| 多形类杆菌 (Bacteroides thetaiotaomicron VPI-5482) | NC_004663 | 84 | 梅毒螺旋体梅毒亚种 (Treponema pallidum subsp. pallidum SS14) | NC_010741 | 32 |
| 鞘脂菌(Sphingobium japonicum UT26S) | NC_014006 | 80 | 梅毒螺旋体 (Treponema paraluiscuniculi Cuniculi A) | NC_015714 | 32 |
| 生丝微菌属(Hyphomicrobium sp. MC1) | NC_015717 | 70 | 鼠疫杆菌(Yersinia pestis CO92) | NC_003143 | 31 |
| 长双歧杆菌(B. longum NCC2705) | NC_004307 | 69 | 台湾贪铜菌 (Cupriavidus taiwanensis LMG 19424) | NC_010528 | 31 |
| 大肠杆菌(E. coli O157:H7 str. Sakai) | NC_002695 | 55 | 噬纤维素菌属 (Cellulophaga algicola DSM 14237) | NC_014934 | 31 |
| 哈维弧菌(V. harveyi ATCC BAA-1116) | NC_009783 | 53 | 结核分枝杆菌(M. tuberculosis H37Rv) | NC_000962 | 30 |
| 溶血葡萄球菌(S. haemolyticus JCSC1435) | NC_007168 | 51 | 沙漠自然球菌(Deinococcus deserti VCD115) | NC_012526 | 30 |
| 缓纤维梭菌 (Clostridium lentocellum DSM 5427) | NC_015275 | 51 | 溃疡拟杆菌(Bacteroides helcogenes P 36-108) | NC_014933 | 27 |
| 表皮葡萄球菌(S. epidermidis RP62A) | NC_002976 | 47 | 金黄色葡萄球菌 (S. aureus subsp. Aureus Mu50) | NC_002758 | 26 |
| 鼠疫杆菌(Y. pestis KIM10+) | NC_004088 | 46 | 金黄色葡萄球菌 (S. aureus subsp. Aureus N315) | NC_002745 | 25 |
| 海单孢菌属 (Marinomonas mediterranea MMB-1) | NC_015276 | 43 | 内脏臭气杆菌 (Odoribacter splanchnicus DSM 20712) | NC_015160 | 25 |
| 红球菌(Rhodococcus jostii RHA1) | NC_008268 | 40 | 嗜热盐碱细菌 (Natranaerobius thermophilus JW/NM-WN-LF) | NC_010718 | 23 |
| 大肠杆菌(E. coli O157:H7 str. EDL933) | NC_002655 | 39 | 圆柱杆菌(Teredinibacter turnerae T7901) | NC_012997 | 23 |
| 固氮密螺旋体(T. azotonutricium ZAS-9) | NC_015577 | 39 | 盐孢菌属(Salinispora tropica CNB-440) | NC_009380 | 20 |
| 白蚁塞巴鲁德氏菌 (Sebaldella termitidis ATCC 33386) | NC_013517 | 38 | 巴西浮霉状菌 (Planctomyces brasiliensis DSM 5305) | NC_015174 | 20 |
| 金黄色葡萄球菌(S. aureus subsp.COL) | NC_002951 | 37 | 苜蓿根瘤菌(Sinorhizobium meliloti AK83) | NC_015590 | 20 |
| 肺炎衣原体 (Chlamydophila pneumoniae AR39) | NC_002179 | 36 |
表2
含有20个以上的假定ORFs被注释上准确功能的基因组的信息"
| 菌株 | NC序列号 | 数量 | 菌株 | NC序列号 | 数量 |
|---|---|---|---|---|---|
| 大肠杆菌(E. coli O111:H-str. 11128) | NC_013364 | 80 | 炭疽芽孢杆菌(B. anthracis str. CDC 684) | NC_012581 | 23 |
| 肠道沙门氏菌(S. enterica subsp. enterica serovar Paratyphi B str. SPB7) | NC_010102 | 64 | 肠道沙门氏菌(S. enterica subsp. enterica serovar Paratyphi C strain RKS4594) | NC_012125 | 23 |
| 鲍氏志贺菌(Shigella boydii CDC 3083-94) | NC_010658 | 52 | 金黄色葡萄球菌(S. aureus subsp. aureus MW2) | NC_003923 | 23 |
| 肠道沙门氏菌(S. enterica subsp. arizonae serovar 62:z4, z23:- str. RSK2980) | NC_010067 | 45 | 蜡状芽孢杆菌(Bacillus cereus ATCC 10987) | NC_003909 | 22 |
| 枯草芽孢杆菌(B. subtilis subsp. subtilis str. 168) | NC_000964 | 42 | 大肠杆菌(E. coli O26:H11 str. 11368) | NC_013361 | 22 |
| 鼠李糖乳杆菌(Lactobacillus rhamnosus Lc 705) | NC_013199 | 38 | 肺炎链球菌(Streptococcus pneumoniae P1031) | NC_012467 | 22 |
| 鼠李糖乳杆菌(L. rhamnosus GG) | NC_013198 | 36 | 金黄色葡萄球菌(S. aureus subsp. aureus Mu3) | NC_009782 | 21 |
| 肠道沙门氏菌(S. enterica subsp. enterica serovar Paratyphi A str. AKU_12601) | NC_011147 | 35 | 金黄色葡萄球菌(S. aureus subsp. aureus JH9) | NC_009487 | 21 |
| 大肠杆菌(E. coli O157:H7 str. Sakai) | NC_002695 | 34 | 结核分枝杆菌(M. tuberculosis CDC1551) | NC_002755 | 20 |
| 大肠杆菌(E. coli O157:H7 str. EDL933) | NC_002655 | 30 | 炭疽芽孢杆菌(B. anthracis str. A0248) | NC_012659 | 20 |
| 肠道沙门氏菌(S. enterica subsp. enterica serovar Paratyphi A str. ATCC 9150) | NC_006511 | 30 | 大肠杆菌(E. coli O157:H7 str. EC4115) | NC_011353 | 20 |
| 大肠杆菌(E. coli SE11) | NC_011415 | 28 | 炭疽芽孢杆菌(B. anthracis str. Ames) | NC_003997 | 20 |
| 大肠杆菌(E. coli ED1a) | NC_011745 | 27 | 哈维氏弧菌(V. harveyi ATCC BAA-1116) | NC_009783 | 20 |
| 大肠杆菌(E. coli UTI89) | NC_007946 | 26 | 结核分枝杆菌(M. tuberculosis KZN 1435) | NC_012943 | 20 |
| 枯草芽孢杆菌(B. subtilis BSn5) | NC_014976 | 25 | 大肠杆菌(E. coli O55:H7 str. CB9615) | NC_013941 | 20 |
| 肠道沙门氏菌 (S. enterica subsp. enterica serovar Typhi str. Ty2) | NC_004631 | 25 | 牛型分枝杆菌 (Mycobacterium bovis AF2122/97) | NC_002945 | 20 |
| 大肠杆菌(E. coli CFT073) | NC_004431 | 23 |
表3
大肠杆菌(E.coli str. K-12 substr. MG1655)满足宽松阈值的新基因"
| 正/负链(在基因组上位置) | 同源序列来源 | 功能 | E值 | 覆盖度 (%) | 一致性 (%) |
|---|---|---|---|---|---|
| 负链(190551~191603) | 肠道沙门菌 (S. enterica subsp. enterica) | 亮氨酸操纵子先导肽 (leu operon leader peptide) | 8e-139 | 79 | 74.01 |
| 负链(2228549~2228758) | 志贺氏菌属(Shigella) | 多药耐药外膜蛋白MdtQ (multidrug resistance outer membrane protein MdtQ) | 1e-27 | 72 | 100 |
| 负链(4322661~4323281) | 志贺氏菌属(Shigella) | Pn转运体膜通道蛋白组分(membrane channel protein component of Pn transporter) | 4e-108 | 77 | 98.74 |
| 负链(4500432~4500791) | 猪布鲁氏杆菌(Brucella suis) | 磷酸乙醇胺转移酶(MULTISPECIES: phosphoethanolamine transferase) | 3e-45 | 69 | 91.57 |
| 正链(1465410~1467950) | 宋内志贺菌(Shigella sonnei) | 包含蛋白质的自转运体结构域 (autotransporter domain-containing protein) | 0.0 | 74 | 87.03 |
| 正链(1470858~1474013) | 福氏志贺氏菌 (Shigella flexneri) | 自转运体外膜β管(MULTISPECIES: autotransporter outer membrane beta-barrel) | 0.0 | 76 | 100 |
| 正链(2070501~2071211) | 双歧杆菌 (Bifidobacterium longum) | GTPase家族蛋白(GTPase family protein) | 4e-129 | 77 | 99.45 |
| 正链(3993850~3994335) | 痢疾志贺氏菌 (Shigella dysenteriae 1617) | CyaX蛋白(CyaX protein) | 9e-67 | 63 | 96.12 |
| 负链(4506626~4506883) | 红树杆菌属 (Mangrovibacter plantisponsor) | 表皮粘着蛋白E (surface-adhesin protein E) | 1e-28 | 98 | 60 |
表4
9个菌株的名称、NC序列号、基因组大小、基因总数和新注释基因的数目"
| 菌株 | NC序列号 | 基因组大小(bp) | 基因总数 | 新基因的数目 |
|---|---|---|---|---|
| 枯草芽孢杆菌(B. subtilis subsp. subtilis str. 168) | NC_000964 | 4215606 | 4175 | 52 |
| 金黄色葡萄球菌(S. aureus subsp. aureus NCTC 8325) | NC_007795 | 2821361 | 2767 | 61 |
| 酿脓链球菌(S. pyogenes SF370) | NC_002737 | 1852441 | 1696 | 104 |
| 流感嗜血杆菌(H. influenzae Rd KW20) | NC_000907 | 1830138 | 1610 | 123 |
| 嗜酸氧化亚铁硫杆菌(A. ferrooxidans ATCC 23270) | NC_011761 | 2982397 | 3147 | 143 |
| 大肠杆菌(E. coli str. K-12 substr. MG1655) | NC_000913 | 4641652 | 4140 | 246 |
| 脑膜炎奈瑟球菌(N. meningitidis MC58) | NC_003112 | 2272360 | 1953 | 279 |
| 炭疽芽孢杆菌(B. anthracis str. Ames) | NC_003997 | 5227293 | 5039 | 418 |
| 肠道沙门氏菌(S. enterica subsp. enterica serovar Typhi str. CT18) | NC_003198 | 4809037 | 4111 | 577 |
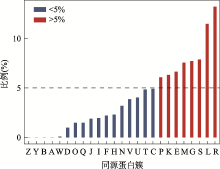
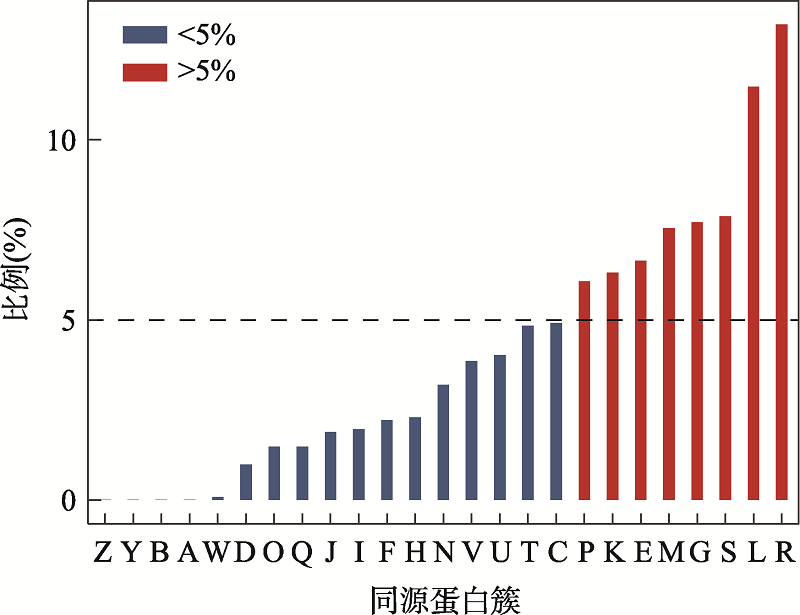
图4
特定同源簇对应的新基因占全部新基因的比例 A:RNA加工和修饰;B:染色质结构和动力学;Y:核结构;Z:细胞骨架;W:细胞外结构;D:细胞周期控制,细胞分裂,染色体分裂;O:翻译后修饰,蛋白反转,伴侣;Q:次生代谢产物的生物合成、运输和分解代谢;I:脂质转运与代谢;J:翻译,核糖体结构和生物发生;H:辅酶运输与代谢;F:核苷酸转运与代谢;V:防卫机制;N:细胞迁移;C:能量产生和转化;U:细胞内传输,分泌和囊泡转运;P:无机离子转运与代谢;T:信号转导机制;K:转录;E:氨基酸转运和代谢;M:细胞壁和细胞膜的生物发生;G:碳水化合物运输和代谢;S:功能未知;R:只能预测大致功能;L:复制重组和修复。"

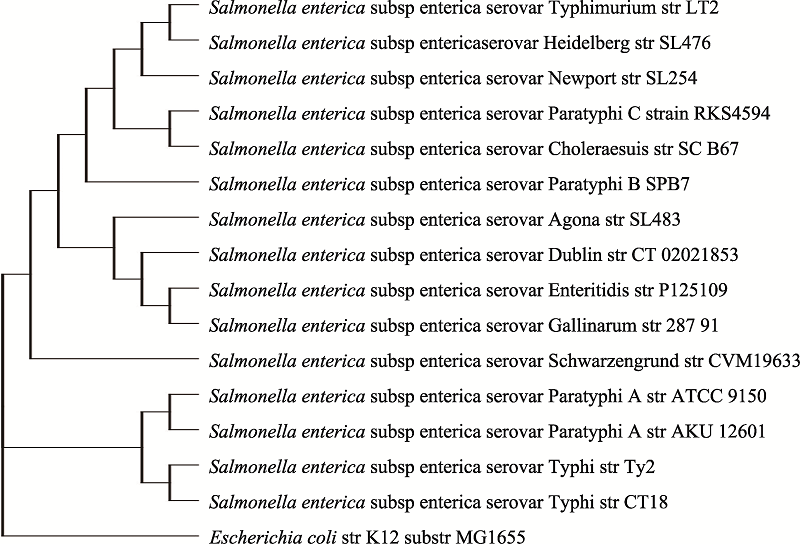
附图1
S. enterica亚种不同血清型的系统发育树"
| [1] | Mørk S, Holmes I . Evaluating bacterial gene-finding HMM structures as probabilistic logic programs. Bioinformatics, 2012,28(5):636-642. |
| [2] | Warren AS, Archuleta J, Feng WC, Setubal JC . Missing genes in the annotation of prokaryotic genomes. BMC Bioinformatics, 2010,11(1):131. |
| [3] | Salzberg SL . Next-generation genome annotation: we still struggle to get it right. Genome Biol, 2019,20(1):92. |
| [4] | Breitwieser FP, Pertea M, Zimin AV, Salzberg SL . Human contamination in bacterial genomes has created thousands of spurious proteins. Genome Res, 2019,29(6):954-960. |
| [5] | Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, Mckenney K, Sutton G, FitzHugh W,Fields C,Gocayne JD,Scott J,Shirley R,Liu LL,Glodek A,Kelley JM,Weidman JF,Phillips CA,Spriggs T,Hedblom E,Cotton MD,Utterback TR,Hanna MC,Nguyen DT,Saudek DM,Brandon RC,Fine LD,Fritchman JL,Fuhrmann JL,Geoghagen NSM,Gnehm CL,McDonald LA,Small KV,Fraser CM,Smith HO,Venter JC. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science, 1995,269(5223):496-512. |
| [6] | Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW . Genbank. Nucleic Acids Res, 2009,37(Database issue):D26-D31. |
| [7] | Yu JF, Xiao K, Jiang DK, Guo J, Wang JH, Sun X . An Integrative method for identifying the over-annotated protein-coding genes in microbial genomes. DNA Res, 2011,18(6):435-449. |
| [8] | Hua ZG, Lin Y, Yuan YZ, Yang DC, Wei W, Guo FB . ZCURVE 3.0: identify prokaryotic genes with higher accuracy as well as automatically and accurately select essential genes. Nucleic Acids Res, 2015,43(W1):W85-W90. |
| [9] | Zickmann F, Renard BY . IPred-integrating ab initio and evidence based gene predictions to improve prediction accuracy. BMC Genomics, 2015,16(1):134. |
| [10] | Keilwagen J, Wenk M, Erickson JL, Schattat MH, Grau J, Hartung F . Using intron position conservation for homology-based gene prediction. Nucleic Acids Res, 2016,44(9):e89. |
| [11] | Besemer J, Lomsadze A, Borodovsky M . GeneMarkS:a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res, 2001,29(12):2607-2618. |
| [12] | Kelley DR, Liu B, Delcher AL, Pop M, Salzberg SL . Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Res, 2012,40(1):e9. |
| [13] | Larsen TS, Krogh A . EasyGene-a prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinformatics, 2003,4(1):21. |
| [14] | Guo FB, Ou HY, Zhang CT . ZCURVE: a new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res, 2003,31(6):1780-1789. |
| [15] | Du MZ, Guo FB, Chen YY . Gene re-annotation in genome of the extremophile Pyrobaculum Aerophilum by using bioinformatics methods. J Biomol Struct Dyn, 2011,29(2):391-401. |
| [16] | Guo FB, Xiong LF, Teng JL, Yuen KY, Lau SK, Woo PC . Re-annotation of protein-coding genes in 10 complete genomes of Neisseriaceae family by combining similarity- based and composition-based methods. DNA Res, 2013,20(3):273-286. |
| [17] | Lei Y, Kang SK, Gao JX, Jia XS, Chen LL . Improved annotation of a plant pathogen genome Xanthomonas oryzae pv. oryzae PXO99A. J Biomol Struct Dyn, 2013,31(3):342-350. |
| [18] | Mao Y, Yang X, Liu Y, Yan Y, Du Z, Han Y, Song Y, Zhou L, Cui Y, Yang R . Reannotation of Yersinia pestis strain 91001 Based on Omics Data. Am J Trop Med Hyg, 2016,95(3):562-570. |
| [19] | Pfeiffer F, Bagyan I, Alfaro‐Espinoza G,Zamora‐Lagos MA,Habermann B,Marin‐Sanguino A,Oesterhelt D,Kunte HJ. Revision and reannotation of the Halomonas elongata DSM 2581T genome. MicrobiologyOpen, 2017,6(4):e00465. |
| [20] | Delcher AL, Bratke KA, Powers EC, Salzberg SL . Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics, 2007,23(6):673-679. |
| [21] | Zhang R, Zhang CT . A Brief Review:The Z-curve theory and its application in genome analysis. Curr Genomics, 2014,15(2):78-94. |
| [22] | Weiss MC, Sousa FL, Mrnjavac N, Neukirchen S, Roettger M, Nelson-Sathi S, Martin WF . The physiology and habitat of the last universal common ancestor. Nat Microbiol, 2016,1(9):16116. |
| [23] | Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ . Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 2010,11(1):119. |
| [24] | Barrett TT, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Edgar R . NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res, 2009,37(Database issue):D885-D890. |
| [25] | Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO,von Mering C. PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteomics, 2012,11(8):492-500. |
| [26] | McGinnis S, Madden TL . BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nuleic Acids Res, 2004,32(Suppl.2):W20-W25. |
| [27] | Wood DE, Lin H, Levy-Moonshine A, Swaminathan R, Chang YC, Anton BP, Osmani L, Steffen M, Kasif S, Salzberg SL . Thousands of missed genes found in bacterial genomes and their analysis with COMBREX. Biol Direct, 2012,7(1):37. |
| [28] | Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walker MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, Jensen LJ, Mering CV, Bork P. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res, 2016,44(Database issue):D286-D293. |
| [29] | Wu ST, Zhu ZW, Fu LM, Niu BF, Li WZ . WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics, 2011,12(1):444. |
| [30] | Tatusov RL, Galperin MY, Natale DA, Koonin EV . The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res, 2000,28(1):33-36. |
| [31] | Qi J, Luo H, Hao BL . CVTree: a phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res, 2004,32:W45-W47. |
| [32] | Hockenbery D, Nuñez G, Milliman C, Schreiber RD, Korsmeyer SJ . Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature, 1990,348(6299):334-336. |
| [33] | Liu WQ, Feng Y, Wang Y, Zou QH, Chen F, Guo JT, Peng YH, Jin Y, Li YG, Hu SN, Johnson RN, Liu GR, Liu SL . Salmonella paratyphi C: Genetic divergence from Salmonella choleraesuis and pathogenic convergence with Salmonella typhi. PLoS One, 2009,4(2):e4510. |
| [34] | Vankuren NW, Long M . Gene duplicates resolving sexual conflict rapidly evolved essential gametogenesis functions. Nat Ecol Evol, 2018,2(4):705-712. |
| [35] | Minor LL, Bockemühl J . 1987 supplement (no.31) to the schema of Kauffmann-White. Ann Inst Pasteur Microbiol, 1988,139(3):331-335. |
| [36] | Dwyer DJ, Belenky PA, Yang JH, Macdonald IC, Martell JD, Takahashi N, Chan CT,lobritz MA,Braff D,Schwarz EG,Ye JD,Pati M,Vercruysse M,Ralifo PS,Allison KR,Khalil AS,Ting AY,Walker GC,Collins JJ. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci USA, 2014,111(20):E2100-E2109. |
| [37] | Hadjeras L, Poljak L, Bouvier M, Morin-Ogier Q, Canal l,Cocaign-Bousquet M,Girbal L,Carpousis AJ. Detachment of the RNA degradosome from the inner membrane of Escherichia coli results in a global slowdown of mRNA degradation, proteolysis of RNase E and increased turnover of ribosome-free transcripts. Mol Microbiol, 2019,111(6):1715-1731. |
| [38] | Kim S, Yu Z, Kil RM, Lee M . Deep learning of support vector machines with class probability output networks. Neural Netw, 2015,64:19-28. |
| [39] | Guo FB . The distribution patterns of bases of protein- coding genes, non-coding ORFs, and intergenic sequences in Pseudomonas aeruginosa PA01 genome and its implication. J Biomol Struct Dyn, 2007,25(2):127-133. |
| [40] | Uyar B, Yusuf D, Wurmus R, Rajewsky N, Ohler U, Akalin A . RCAS: an RNA centric annotation system for transcriptome-wide regions of interest. Nucleic Acids Res, 2017,45(10):e91. |
| [41] | Huang Y, Liu Q, Chi LJ, Shi CM, Wu Z, Hu M, Shi H, Chen H . Application of BIG-Annotator in the genome sequencing data functional annotation and genetic diagnosis. Hereditas(Beijing), 2018,40(11):1015-1023. |
| 黄莹, 刘琪, 池连江, 石承民, 吴祯, 胡敏, 石宏, 陈华 . BIG-Annotator: 基因组测序数据高效功能注释及其在遗传诊断中的应用. 遗传, 2018,40(11):1015-1023. | |
| [42] | Bick JT, Zeng SQ, Robinson MD, Ulbrich SE, Bauersachs S . Mammalian Annotation Database for improved annotation and functional classification of Omics datasets from less well-annotated organisms. Database(Oxford), 2019,2019:1-16. |
| [43] | Ravindran SP, Lüneburg J, Gottschlich L, Tams V, Cordellier M . Daphnia stressor database: Taking advantage of a decade of Daphnia ‘-omics’ data for gene annotation. Sci Rep, 2019,9(1):11135. |
| [1] | 文钟灵, 杨旻恺, 陈星雨, 郝晨宇, 任然, 储淑娟, 韩洪苇, 林红燕, 陆桂华, 戚金亮, 杨永华. 酸铝胁迫土壤中耐铝大豆根际不同部位细菌群落结构、功能及其对促生菌富集作用的研究[J]. 遗传, 2021, 43(5): 487-500. |
| [2] | 李金玉, 杨姗, 崔玉军, 王涛, 滕越. 细菌最小基因组研究进展[J]. 遗传, 2021, 43(2): 142-159. |
| [3] | 高志伟, 王龙. 真核生物起源研究进展[J]. 遗传, 2020, 42(10): 929-948. |
| [4] | 杨超, 杨瑞馥, 崔玉军. 细菌全基因组关联研究的方法与应用[J]. 遗传, 2018, 40(1): 57-65. |
| [5] | 谢龙祥, 于召箫, 郭思瑶, 李萍, AbualgasimElgailiAbdalla, 谢建平. 表观遗传和蛋白质翻译后修饰在细菌耐药中的作用[J]. 遗传, 2015, 37(8): 793-800. |
| [6] | 李志芳, 冯自力, 赵丽红, 师勇强, 冯鸿杰, 朱荷琴. 转几丁质酶和葡聚糖酶双价基因棉花对土壤细菌种群多样性的影响[J]. 遗传, 2015, 37(8): 821-827. |
| [7] | 杨小亮,白大章,邱巍,董慧琴,李大全,陈芳,马润林,Hugh T Blair,高剑峰. 以绵羊MHC区段BAC克隆酶切片段为探针杂交筛选绵羊混合组织cDNA文库[J]. 遗传, 2012, 34(7): 887-894. |
| [8] | 谢池楚,贾海云,陈月华. 细菌几丁质酶基因的表达调控[J]. 遗传, 2011, 33(10): 1029-1038. |
| [9] | 谢兆辉. RNA沉默在植物生物逆境反应中的作用[J]. 遗传, 2010, 32(6): 561-570. |
| [10] | 刘关君,王丽娟,秦智伟,孟令波 . 黄瓜叶片细菌性角斑病侵染初期cDNA文库分析[J]. 遗传, 2009, 31(10): 1042-1048. |
| [11] | 任洪林,徐丹丹,乔琨,蔡灵,黄伟滨,张鼐,王克坚. 细菌攻毒杂色鲍血淋巴细胞抑制性差减杂交文库构建及巨噬细胞表达蛋白cDNA的克隆与差异表达[J]. 遗传, 2008, 30(8): 1043-1050. |
| [12] | 裔传灯,龚志云,梁国华,王飞华,汤述翥,顾铭洪. 稻属不同染色体组着丝粒BAC克隆的分离和鉴定[J]. 遗传, 2007, 29(7): 851-858. |
| [13] | 徐晓红,吴敏,张会斌,刘志虎. 嗜盐古生菌br基因的遗传分析[J]. 遗传, 2007, 29(3): 376-376―380. |
| [14] | 杨,军,尹启生,侯明生. 植物病原细菌的hrp基因[J]. 遗传, 2005, 27(5): 852-858. |
| [15] | 陈师勇,莫照兰,张振冬,邹玉霞,徐永立,张培军. 细菌毒力基因体内表达检测技术研究进展[J]. 遗传, 2005, 27(3): 505-511. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||

www.chinagene.cn
备案号:京ICP备09063187号-4
总访问:,今日访问:,当前在线:
